










Iron deficiency is one of the most common nutritional deficits, affecting more than 2 billion people worldwide.1 Iron deficiency often occurs in children, women, elderly people and patients with chronic inflammatory conditions.1 Iron is an essential element in enzymes and proteins involved in mitochondrial function, oxygen transport and biodegradation and the functioning of proteins.2 The body has no pathway of actively excreting iron and its metabolism is mainly regulated through the intestinal uptake of iron or the buffering of iron within cells.
In the bloodstream, iron is bound to transferrin, which is capable of binding two Fe3+ ions. Intracellularly, iron is bound to ferritin which is capable of binding numerous iron ions.3 Because ferritin carries most of the body’s iron and is also secreted in the blood stream, this reliably reflects a total body iron content in conditions without inflammation. The rate-dependent step in iron uptake is located at the level of ferroportin, which is a protein present at the basolateral site of the enterocyte and is present on spleen macrophages. The expression of this protein is regulated through hepcidin. Hepcidin is upregulated in conditions of inflammation, such as heart failure and excess iron, while it is downregulated in the presence of an iron deficit or hypoxaemia. Hepcidin results in an internalisation of ferroportin, leading to less uptake of intestinal iron from the gut, but also clustering of iron within the reticulo-endothelial system.4 This aspect of iron metabolism is important to appreciate because it explains why a functional deficit of iron can occur in heart failure due to reticulo-endothelial system clustering, and why oral iron therapy is inefficient in the setting of heart failure because of the degradation of ferroportin in enterocytes which hampers intestinal uptake.
Numerous mechanisms result in the high prevalence of iron deficiency in heart failure. Iron deficiency might result from enhanced iron loss, such as from blood loss in patients on antiplatelets or anticoagulants, diminished iron uptake because of malnutrition, intestinal congestion or tea drinking, or reduced iron bio-availability because of reticulo-endothelial clustering.
As iron plays such an important role in numerous important physiological pathways, its presence can worsen the disease trajectory of the patient with heart failure. This review focuses on the role of iron deficiency in aggravating cardiac function and structure and the mitigating role treatment with ferric carboxymaltose plays in patients with heart failure with reduced ejection fraction (HFrEF).5
Definition and Prevalence of Iron Deficiency
Heart failure is associated with chronic inflammation. Ferritin is an acute phase protein, so its levels are elevated in the setting of heart failure. Therefore, iron deficiency can be diagnosed even in the face of higher levels of ferritin. No formal validation studies have established a definition of iron deficiency in heart failure.6,7 Yet, findings from other medical fields, such as nephrology and haematology, have been used to formulate a definition of iron deficiency in heart failure.
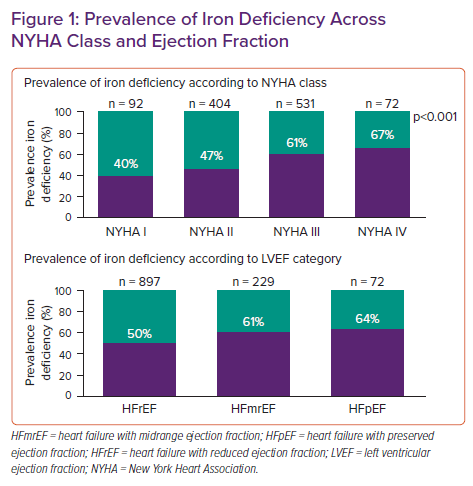
The current established definition of iron deficiency in heart failure is a ferritin level below 100 ng/ml irrespective of transferrin saturation (TSAT) or ferritin between 100–300 ng/ml if TSAT is <20%. Importantly, this definition of iron deficiency is capable of identifying heart failure patients with more advanced symptoms, poor quality of life (QoL), diminished exercise capacity and a higher risk of hospitalisation due to heart failure.8–13 Using this definition in intervention trials of IV iron has allowed the identification of patients who show improvement in terms of functional status, exercise capacity and clinical outcome after treatment with ferric carboxymaltose.14–17 This underscores the usefulness of the definition, despite no initial formal validation against the gold standard bone marrow iron staining. Using this definition in patients with stable heart failure, about 50% of patients will be diagnosed with iron deficiency, a prevalence that has been found in numerous observational studies.8,9,11,13,18
Figure 1 shows the prevalence of iron deficiency according to left ventricular ejection fraction (LVEF) strata showing a high prevalence in heart failure with reduced (HFrEF), mildly reduced (HFmrEF) and preserved (HFpEF) ejection fraction.18 The prevalence of iron deficiency is also higher in patients with a higher New York Heart Association (NYHA) heart failure class. Similarly, in acute heart failure there is a higher prevalence of iron deficiency with about 80% of patients being affected.19 Yet, 30 days after a hospital admission for acute heart failure this prevalence significantly drops, illustrating the difficulty of diagnosing iron deficiency in acute heart failure.20 However, the AFFIRM-AHF trial shows that in the setting of acute heart failure, the classic diagnostic criteria are capable of identifying patients with iron deficiency who benefit from treatment with ferric carboxymaltose showing that this definition of iron deficiency remains useful in selecting patients even in the setting of acute heart failure.16
Clinical Impact of Iron Deficiency
Given the wide range of biological roles that iron plays, it is not surprising that iron deficiency is associated with numerous clinical manifestations. Iron is an essential co-factor in the first three elements of the electron transport chain and is associated with diminished oxidative phosphorylation and reduced intracellular content of phosphocreatinine and adenosine triphosphate (ATP), which will manifest in tissues with high energetic demand, such as the myocardium, skeletal muscle and the central nervous system. Patients with iron deficiency often report having a lack of energy, reduced QoL, headaches, limited concentration, reduced exercise capacity and shortness of breath.18
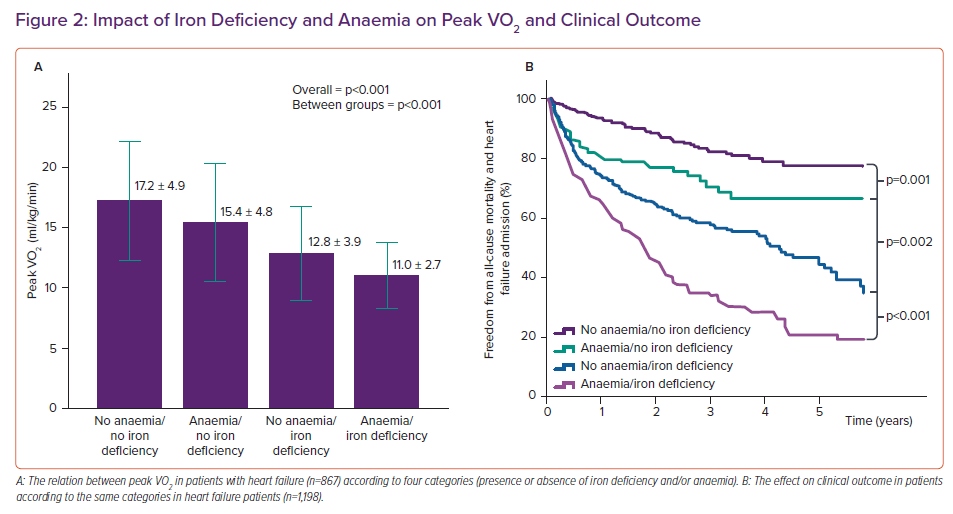
Furthermore, heart failure is associated with a higher risk of hospitalisation for heart failure or dying from a cardiovascular cause. Even in the absence of anaemia, iron deficiency is associated with a reduced exercise capacity and worse clinical outcome as reflected in Figure 2 which also shows that anaemia due to other reasons than iron deficiency has less of an impact on exercise capacity and clinical outcome than iron deficiency does in the absence of anaemia.18
The Impact of Iron Deficiency on Cardiac Function
According to the Fick equation, peak VO2 is determined by peak exercise cardiac output and the arterial minus mixed venous O2 content (arteriovenous oxygen difference; A-VO2), with the latter being dependent on haemoglobin levels and peripheral oxygen extraction. Theoretically, iron deficiency could affect all elements of the Fick equation while anaemia mainly affects the arterial O2 content component, explaining why isolated iron deficiency (without anaemia) has a stronger relation with peak VO2 than anaemia unrelated to iron deficiency.10,18
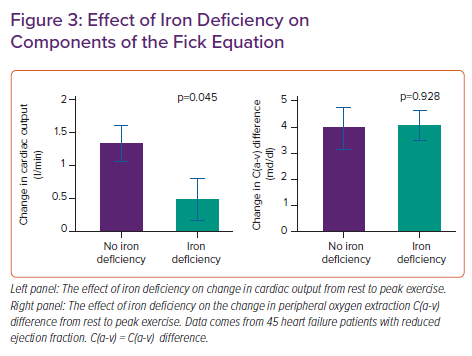
We performed a haemodynamic study using invasive cardiopulmonary exercise testing (iCPET) in heart failure patients with reduced ejection fraction.21 We showed that patients with iron deficiency had less ability to increase their cardiac output compared with patients without iron deficiency (see Figure 3). The A-VO2 difference was similar between patients with or without iron deficiency, indicating a similar degree of skeletal muscle oxygen extraction. Nevertheless, the role of iron deficiency in affecting skeletal muscle has been well documented indicating early muscle acidification, lower energy content and slowed recovery kinetics; however, this spans beyond the scope of this article.22–24 Additionally, a similar degree of A-VO2 difference for a lower attained cardiac output could be regarded as abnormal according to the Fick principle of diffusion. This is because a lower cardiac output should be met with a higher A-VO2 difference due to the slower capillary passing time where there is more time to extract oxygen. Therefore, our study points to both cardiac muscle and skeletal muscle involvement in the pathophysiology of iron deficiency.22–24
Cellular Effect of Iron Deficiency
It is important to appreciate the effect of iron deficiency at a cellular level to understand the impact of iron deficiency on the macroscopic cardiovascular system. On a cellular level it is important to remember the intertwining of cellular excitation contraction coupling of cardiomyocytes and energy metabolism.
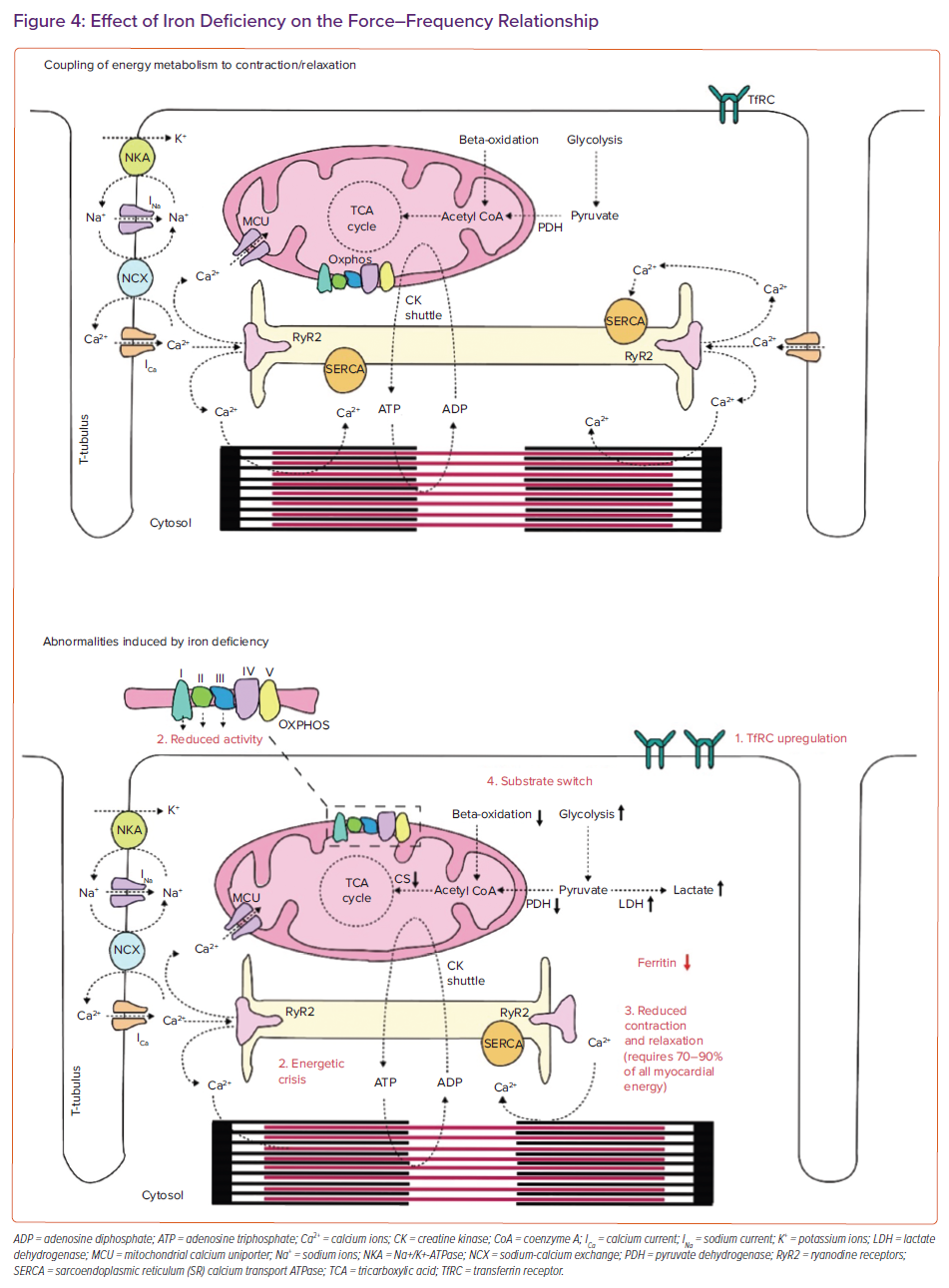
More than any muscle, the heart consumes ATP with an astonishing rate of 6 kg a day.25 Most myocardial energy is required for either myofilament shortening (the cellular basis of systole) or calcium buffering (the cellular basis of diastole). Once myocytes become activated by a depolarising sodium current, this results in cellular calcium influx from both the extracellular compartment and the endoplasmic reticulum (Figure 4). Such calcium influx results in actin myosin overlap and myofilament shortening. However, some cytoplasmic calcium is taken up by the mitochondria via the mitochondrial calcium uniporter (MCU).26 Within the mitochondria, calcium plays a role in determining the rate of substrate use in the tricarboxylic acid (TCA) cycle, which is coupled to the rate of oxidative phosphorylation in the electron transport chain (because the TCA cycle provides reducing equivalents). As a result, calcium cycling is fine-tuned to the rate of ATP production. In the cell, ATP is converted to phosphocreatine which is shuttled in the subcellular space through creatinine kinase isoforms to regions of high energy demand, being the myofilaments for shortening or the sarcoendoplasmic reticulum calcium transport ATPase pump for calcium buffering. In this way, excitation coupling is fine-tuned to energy metabolism, forming a streamlined and efficient engine (Figure 4).
Hoes et al. explored the role of iron deficiency on myocyte energy metabolism and myocyte shortening and relaxation.27 Human embryonic stem cell-derived cardiomyocytes were incubated with an iron chelator (deferoxamine), resulting in a decrease in intracellular iron content. Iron depletion affected mitochondrial function through reduced activity of the iron–sulphur cluster containing complexes I, II and III, but not complexes IV and V, leading to reduced ATP-linked respiration and respiratory reserve.27,28 This energetic crisis resulted in diminished myocyte shortening and slowed and diminished myocyte relaxation kinetics. As can be expected, myocyte iron deficiency resulted in upregulation of membrane-bound transferrin receptors. Interestingly, administration of soluble transferrin-bound iron allowed for a recovery of the morphological and functional consequences induced by iron deficiency. This indicates that the goal of iron therapy should be to replenish transferrin, which will subsequently lead to intracellular iron repletion. As alluded to in the introduction, this is something that can only be achieved by IV rather than iron taken orally, given the reduced intestinal uptake in the setting of heart failure.17,29
Cardiac Performance and Iron Deficiency
While the Hoes et al. study gives a cellular understanding of the consequences of iron deficiency, it needs to be recognised that the model used is an extreme form of myocardial iron deficiency. However, lesser degrees of iron deficiency might still become relevant in clinical practice. Indeed, human embryonic stem cell-derived cardiomyocytes contract at a baseline rate of around 30–35 BPM, something known to the clinician as the rate of idioventricular rhythm. Yet the myocardial energy requirement increases at higher heart rates. Indeed one of several operating mechanisms resulting in an increased cardiac contractility during exercise is the force-frequency relationship.30 This is also known as the Bowditch–Treppe phenomenon and it indicates that at higher heart rates, the myocardium contracts with increased force to meet the enhanced contractile requirements to support the increase in stroke volume during exercise. While such a phenomenon plays in healthy people, as they exhibit a positive force–frequency relationship, illustrated by an increase in force at higher heart rates, this mechanism is deranged in a state of heart failure. Indeed, it has long been recognised that heart failure patients manifest with the reverse, being a negative force-frequency relationship or a decrease in force at higher heart rates.31
The decrease in cardiac force at higher heart rates in patients with heart failure is partially the result of a cellular energetic deficit. Haddad et al. described a mouse model of iron deficiency assessing the impact of iron deficiency on the force-frequency relationship after stimulation with dobutamine.32 Iron-deficient mice showed a similar increase in heart rate to mice without iron deficiency, yet the force (measured invasively as Dp/dt) generated dropped at higher heart rates in iron-deficient mice. Using in vivo 31P magnetic resonance spectroscopy, the authors showed that this negative force-frequency relationship was caused by an energetic crisis induced by iron deficiency as documented by a drop in the phosphocreatine/ATP ratio.32
We have previously shown that a similar principle occurs in heart failure patients with iron deficiency. By prospectively selecting HFrEF patients with a CRT device, we were able to study the force-frequency relationship in vivo.33 By a stepwise increase in the lower rate of biventricular pacing and simultaneously measuring the non-invasive cardiac contractility index (systolic blood pressure/left ventricular end systolic volume index), we showed that iron-deficient heart failure patients exhibit a negative force-frequency relationship. Taken together with the invasive iCPET data showing a lower cardiac output during exercise, this clearly documents that iron deficiency negatively affects cardiac performance, especially during exercise or higher heart rates.21,23,33
The Impact of Iron Deficiency on Cardiac Structure
As well as being a co-factor in the first three elements of the mitochondrial electron transport chain, iron is also an essential co-factor in anti-oxidative enzymes.28 Insufficient myocardial capability to defend against oxidative stress has been implicated in the process of progressive cardiac remodelling forming one of the hallmark features of HFrEF.34 Indeed, animal models of iron deficiency (induced by either knockout or with low iron diets) show progressive cardiac remodelling consisting of ventricular dilatation, ventricular hypertrophy, systolic dysfunction and cardiomyocyte apoptosis thereby implicating iron in the process of progressive cardiac remodelling.35,36 In humans, an approach that also clearly documented the involvement of iron deficiency on cardiac remodelling was shown by studying the modulating role of iron deficiency on cardiac reverse remodelling after CRT implant. CRT as a non-pharmacological treatment option for HFrEF patients with electromechanical dyssynchrony can be seen as a clean model allowing a way to study the influences of a covariate on the process of reverse remodelling.
In comparison to drug interventions, which have numerous extra-cardiac effects, CRT selectively induces cardiac reverse remodelling. Before treatment with IV iron was mainstream, we and several other groups showed in a cohort of historically implanted CRT patients, that those with iron deficiency at the time of CRT implantation showed less cardiac reverse remodelling documented by a persistently reduced LVEF and more dilated ventricle.37,38 These studies implicate iron deficiency in the process of cardiac remodelling. However, these observational studies do not resolve the issue of whether iron deficiency directly stimulates cardiac remodelling or both iron deficiency and cardiac remodelling are a sign of another overarching process, such as inflammation.
Effect of Ferric Carboxymaltose on Cardiac Function and Structure
The causal effect of iron deficiency on cardiac function and structure can only be adequately studied in prospective double blind randomised controlled trials (RCTs), as such studies can establish causation. Previous trials have documented the effect of ferric carboxymaltose on patients’ functional status and submaximal exercise capacity shown in the six-minute walk test both at short and long-term follow-up.14,15 Other trials have shown the beneficial effect on maximal exercise capacity and risk for heart failure admissions.16,17 Yet, few randomised studies have assessed the effect of ferric carboxymaltose on cardiac function and structure. Previous small studies using a different IV iron formulation (iron sucrose) suggested that treatment with IV iron could induce reverse remodelling as documented by an improvement in LVEF.39,40
The Myocardial-IRON trial investigated whether treatment with ferric carboxymaltose is capable of replenishing myocardial iron content.41 Using the feature of the T2* sequence of MRI, investigators estimated myocardial iron content. T2* is often used in clinical practice to examine iron loading, such as for patients undergoing frequent transfusions, with low values indicating myocardial iron loading. However, the reverse has also shown to be true and patients with iron deficiency exhibit an elevated T2* value. In the Myocardial-IRON trial, treatment with ferric carboxymaltose was capable of replenishing myocardial iron content, indicated as a significant decrease in T2* at the 7- and 30-day follow-up. In a post hoc analysis authors also showed that it was associated with an improvement in biventricular function.42
To determine the effect of ferric carboxymaltose on cardiac function and structure, we designed the IRON-CRT trial. In this double blind prospective RCT we included HFrEF patients who were optimally treated with guideline-directed medical therapy and device-based therapies (CRT), but had a persistently reduced LVEF and had iron deficiency. The CRT device in IRON-CRT needed to work adequately documented by more than 6 months ≥98 biventricular pacing. HFrEF patients with a CRT device were specifically chosen because we are able to assess the force-frequency relationship in these patients. Patients were randomised in a double-blind fashion to either standard of care (IV placebo) or ferric carboxymaltose.43
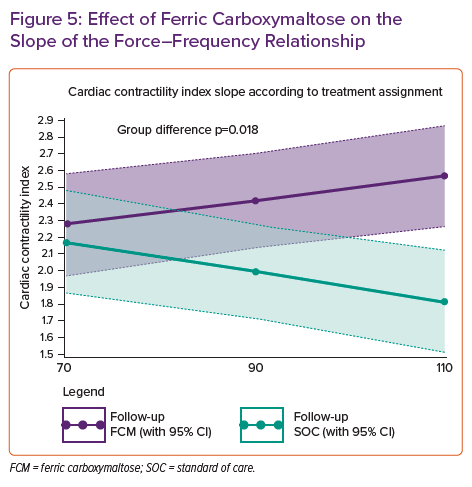
Cardiac structure was assessed as the change in LVEF, left ventricular end systolic volume (LVESV) and end diastolic volume (LVEDV) from baseline to three months follow-up using 3D echocardiography. The latter is important as in well selected patients, 3D echocardiography has the same variability for LV volumetric measurements as MRI. At baseline and follow-up, patients underwent a detailed force-frequency pacing protocol, in which the non-invasive contractility index was measured at 70, 90 and 110 beats of biventricular pacing, allowing us to determine the slope of the force-frequency relationship. At 3 months follow-up, the least square mean change in LVEF from baseline was significantly higher in the ferric carboxymaltose group (4.22%, 95% CI [3.05–5.38]) in comparison to the standard of care group (−0.23%, 95% CI [−1.44, 0.97], p<0.001). Additionally, the change in LVESV from baseline to follow-up was more pronounced in the ferric carboxymaltose group versus the standard of care group (−9.72 ml, 95% CI [−13.5, 5.93] versus −1.83 ml, 95% CI [−5.7, 2.1], p=0.001) However, the change in LVEDV was not different between the two treatment groups (ferric carboxymaltose: −2.5 ml, 95% CI [−5.3, 0.3] versus −1.9 ml, 95% CI [−4.7, 1.0], p=0.784).43 Additionally, treatment with ferric carboxymaltose was capable of improving the force-frequency relationship as documented in Figure 5. In patients treated with ferric carboxymaltose the negative force-frequency relationship was transformed into a positive force-frequency relationship. Something that was not observed in patients who were in the standard of care group. This data, together with the pre-existing knowledge support the notion that through an improvement in cardiac energy reserve the failing ventricle becomes more efficient and is capable of manifesting reverse cardiac remodelling.
Guidelines for Treating Iron Deficiency
The 2021 ESC guidelines for the diagnosis and treatment of heart failure gave a IIa recommendation supporting the role of ferric carboxymaltose in patients with iron deficiency in order to alleviate heart failure symptoms, improve exercise capacity and QoL in patients with an LVEF <45% (which is a slight change in comparison to the 2016 guidelines which defined HFrEF by an LVEF <40%).5 Additionally, a IIa recommendation is given based on the AFFIRM-HF trial supporting the use of ferric carboxymaltose in patients with a recent heart failure admission and an LVEF <50% to reduce the risk for heart failure admissions. Patients with heart failure need to be periodically screened for the presence of iron deficiency according to the 2021 guidelines (class 1c recommendation) because iron deficiency can reoccur. This is also a slight change in comparison to the 2016 guidelines, which recommended screening for the presence of iron deficiency only in the setting of a new diagnosis of heart failure.44
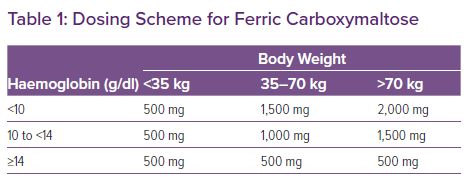
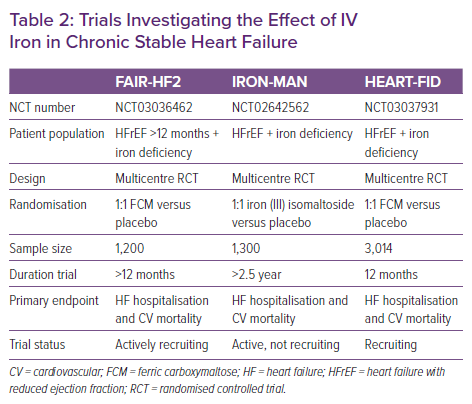
From a treatment perspective, it is important to adhere to the doses of ferric carboxymaltose as used in the RCTs (and formulated in the summary of product characteristics of ferric carboxymaltose), which is based on the haemoglobin levels and the weight of the patient (Table 1). Indeed, treatment with a too low dose is associated with less improvement in functional status.45 Ongoing studies in patients with stable heart failure will further determine the effect of treatment with IV iron on hard clinical endpoints such as cardiovascular mortality and the risk of hospitalisation for heart failure. Table 2 lists ongoing trials with IV iron in the setting of chronic heart failure.
Conclusion
Iron deficiency alters myocyte and myocardial function and structure. Data from smaller mechanistic studies and larger RCTs demonstrate a beneficial effect of treatment with ferric carboxymaltose on cardiac function and structure. These mechanistic observations explain the beneficial effect of ferric carboxymaltose on functional status, exercise capacity and the risk of being hospitalised for heart failure.