










A 46-year-old man presented with progressive dyspnoea and orthopnoea. His past medical history included a history of pulmonary sarcoidosis, end-stage renal disease on haemodialysis, obstructive sleep apnoea and prior ventricular tachycardia with recent ICD firing in the past 6 months. Of note, the patient had been diagnosed with pulmonary sarcoidosis many years prior to presentation; he had initially been treated with prednisone, but had been off any therapy for several years prior to presentation. On presentation, the patient was afebrile with blood pressure 77/58 mmHg, heart rate 70 BPM (atrioventricular paced), respiratory rate 22 breaths/min and oxygen saturation 94% on room air. On physical examination, the patient was lethargic, but responsive to questions and interactive. He had clear lungs, elevated jugular venous pressure of 12 cmH2O and cool, non-oedematous extremities. Laboratory results were notable for sodium 134 mmol/l, potassium 4.8 mmol/l, creatinine 14 mg/dl, troponin I 0.18 ng/ml, brain natriuretic peptide 9,381 pg/ml and lactic acid 0.6 mEq/l. A chest radiograph showed bibasilar opacities, pulmonary vascular congestion and cardiomegaly. Given persistent hypotension and concern for cardiogenic shock, the patient was started on dobutamine and low-dose noradrenaline.
A transthoracic echocardiogram showed severe biventricular dysfunction with marked four-chamber enlargement and moderate to severe tricuspid regurgitation. Right heart catheterisation demonstrated right atrial pressure 34 mmHg, pulmonary artery pressure 48/36 mmHg (mean 38 mmHg), pulmonary capillary wedge pressure 32 mmHg and a cardiac index (Fick) of 1.87 l/min/m2. Coronary angiography demonstrated non-obstructive coronary artery disease suggestive of a non-ischaemic cardiomyopathy as the aetiology of the patient’s heart failure. An intra-aortic balloon pump (IABP) was placed for additional support, and continuous renal replacement therapy was initiated for haemodialysis and volume removal. Thereafter, the patient’s haemodynamics improved, allowing for weaning and removal of IABP support, transition to intermittent haemodialysis and transfer to the step-down unit on dobutamine support.
Although the differential for the patient’s non-ischaemic cardiomyopathy was broad, our leading diagnosis was cardiac sarcoidosis (CS), given his history of pulmonary sarcoidosis and ventricular tachycardia. Imaging studies were performed for further characterisation of the patient’s cardiomyopathy while he underwent evaluation for dual heart and kidney transplantation.
Investigations
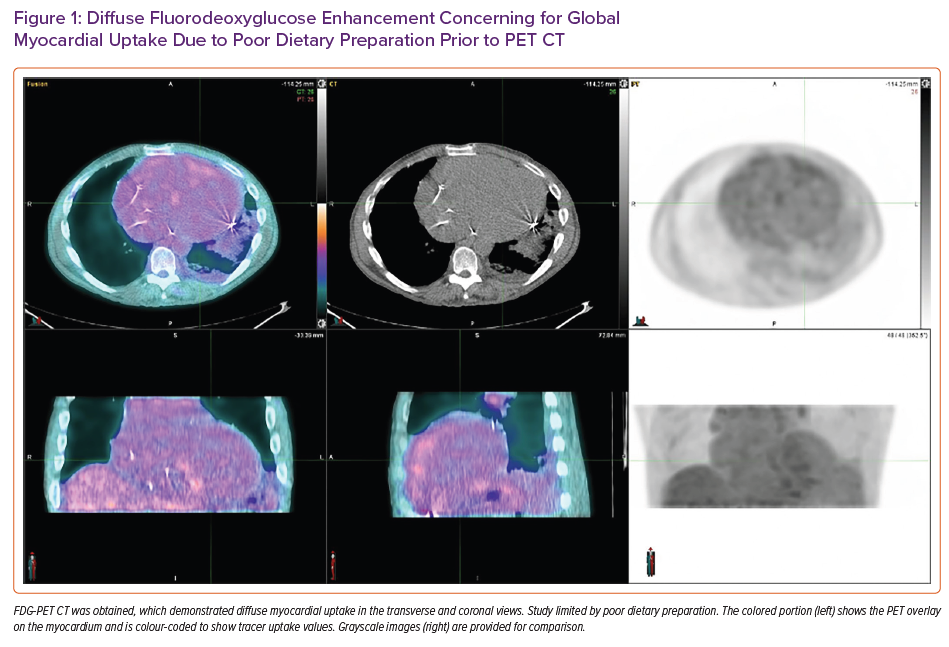
Fluorodeoxyglucose (FDG)-PET CT showed diffuse areas of increased FDG uptake involving the anterior, anteroseptal and basal inferoseptal left ventricular walls and right ventricular free wall (Figure 1). The study was limited by poor dietary preparation and was rendered non-diagnostic for active CS.
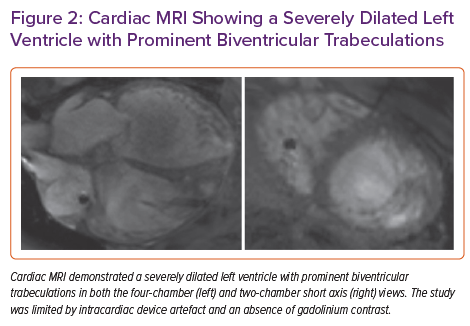
Cardiac MRI without contrast showed prominent intracavitary trabeculations and was diagnostic of left ventricular non-compaction (LVNC) cardiomyopathy. The end-diastolic non-compacted to compacted myocardium diameter ratio was 2.5 (Figure 2). Prominent trabeculations were also noted in the right ventricle. In the absence of contrast and parametric mapping, the latter of which was impaired by ICD artefact, cardiac MRI was non-diagnostic for infiltrative cardiomyopathies.
Despite continued dobutamine support, the patient experienced recurrent cardiogenic shock in the ensuing days and was transferred back to the intensive care unit for reinsertion of the IABP. He was approved for dual heart and kidney transplantation. After 7 days, the patient underwent combined organ transplant.
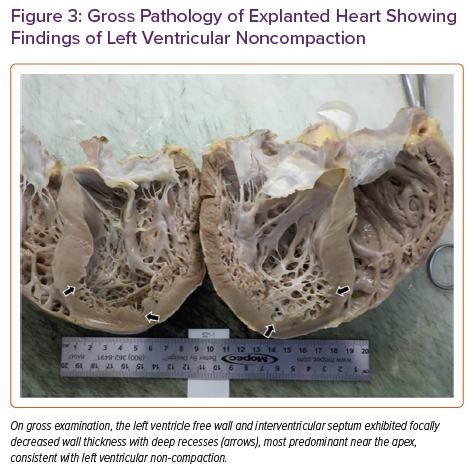
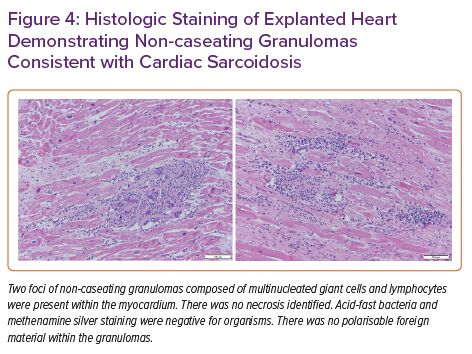
Pathology of the explanted heart showed focal non-compaction of the left ventricular free wall with prominent biventricular trabeculations (Figure 3). Haematoxylin and eosin staining of the left ventricular free wall, interventricular septum (Figure 4) and right ventricle showed non-caseating granulomas consistent with CS. The patient had a post-transplant course complicated by recurrent gastrointestinal bleeding requiring colectomy, but was ultimately successfully discharged to inpatient rehabilitation and then back home.
Discussion
We present an interesting case of a 46-year-old man with known pulmonary sarcoidosis presenting with undifferentiated cardiomyopathy leading to eventual orthotopic heart transplantation. Cardiac MRI prior to transplantation was suspicious for non-compaction cardiomyopathy; however, myocardial sarcoidosis could not be excluded non-invasively due to poor PET quality and intracardiac device artefact. Gross examination of the explanted heart after transplant showed evidence of both non-compaction and sarcoidosis. There have been two prior case reports of the co-occurrence of non-compaction and sarcoidosis in patients with cardiomyopathy. 1,2 However, this is the first report to confirm the presence of both disorders by gross pathology and histology.
Sarcoidosis is a multi-organ inflammatory disease of uncertain aetiology resulting in the formation of non-caseating granulomas and scar. Although 5% of patients with systemic sarcoidosis develop clinically significant CS, autopsy studies suggest that approximately 25% of cases involve the heart.3 When present, CS can cause conduction system abnormalities, ventricular and supraventricular tachyarrhythmias, severe heart failure and sudden cardiac death.4 A small percentage of patients with CS will have progressive heart failure or arrhythmias ultimately requiring implantation of a left ventricular assist device or cardiac transplantation. Diagnosis of CS in end-stage cardiomyopathy remains critically important in patients referred for transplantation; confirmed CS may inform post-transplant immunosuppression strategies to limit recurrence. However, post-transplant recurrence of CS appears to be uncommon, and post-transplant outcomes are similar to those of non-CS patients.5
The diagnosis of CS remains challenging due to the lack of standardised clinical criteria. Although endomyocardial biopsy with confirmed histopathological evidence of non-caseating granulomas is the gold standard, it is limited by poor sensitivity, with a diagnostic yield of only approximately 25% on unguided biopsy.6 As such, many clinical guidelines rely on histological diagnosis of extracardiac sarcoidosis, along with clinical findings of cardiac involvement, although this does not account for the cohort of patients with isolated CS.6 However, detection has improved over time with advances in non-invasive imaging, including FDG-PET and cardiac MRI with multiparametric mapping. Both imaging modalities play a vital role in the assessment of CS. Cardiac MRI has emerged as a valuable tool in assisting with both the diagnosis and risk stratification of patients with CS. For example, cardiac MRI can help predict the risk of life-threatening arrhythmia based on the burden of late gadolinium enhancement (LGE). An association between the degree of LGE on cardiac MRI and poor prognosis has been shown in several studies.4,7–9 Although sensitive for the diagnosis of CS, cardiac MRI is less sensitive in the detection of active inflammation, thus limiting its usefulness when guiding immunosuppressive therapy. FDG-PET can also be used to aid in the diagnosis and prognostication of CS, and has the additional advantage of measuring the response to immunosuppressive therapy. Thus, the use of PET and cardiac MRI is complementary and, in conjunction, these imaging modalities improve the diagnosis and treatment of CS.
Although advances in cardiac imaging have aided in the diagnosis of CS, cardiac imaging is not without limitations. A retrospective study by Divakaran et al. looked to correlate pretransplant imaging findings of likely CS with post-transplant histological findings from explanted hearts.10 In that study, among the 205 patients who received heart transplants during the study period, 18 underwent FDG-PET and 31 underwent cardiac MRI prior to transplant. Patients were then categorised by likelihood of CS diagnosis based on FDG-PET and cardiac MRI findings. Of the five cases considered highly probable by FDG-PET, all five were confirmed as CS on pathology and all had evidence of extracardiac FDG uptake on their examination. However, of the nine patients with probable CS by FDG-PET, only one was confirmed as CS on post-transplant pathology.10 This suggests that although FDG-PET is highly sensitive for the diagnosis of CS, its specificity decreases in the absence of extracardiac findings and high pretest probability of disease. The cardiac MRI findings were difficult to interpret because only one of 31 patients in the cardiac MRI group had confirmed CS. In our case, the patient underwent both cardiac MRI and FDG-PET prior to heart transplantation. However, both studies were non-diagnostic for CS, despite post-transplant histopathological confirmation. Cardiac MRI in our patient was limited by the lack of contrast administration. Imaging criteria in the study of Divakaran et al. relied on LGE patterns to determine the likelihood of CS.10 Thus, in the absence of gadolinium enhancement, the use of cardiac MRI to diagnose CS is limited. The use of FDG-PET in our patient was limited by poor dietary preparation.
LVNC, also known as left ventricular hypertrabeculation, is a myocardial structural abnormality of unclear aetiology. LVNC is characterised structurally by a spongy endocardial layer and a thinner, compacted epicardial layer of the myocardium at the left ventricular lateral wall and apex.11 Although the exact aetiology is unknown, it is thought that hypertrabeculation results from a failure of the myocardial compaction process during normal cardiac development.12 Although usually asymptomatic, it can be complicated by heart failure, thrombus formation, malignant ventricular tachyarrhythmias and sudden cardiac death. Non-compaction cardiomyopathy can cause both systolic and diastolic dysfunction. Although the cause of systolic dysfunction in LVNC is unclear, there is some evidence suggesting that it may be due to subendocardial hypoperfusion.1 Diastolic dysfunction in LVNC is felt to be related to abnormal relaxation secondary to hypertrabeculation.11 Imaging studies, such as echocardiography and cardiac MRI, are primarily used to diagnose LVNC, although at present there is no consensus regarding the exact diagnostic criteria. The prevalence of LVNC is reported to be quite low, but it may be under-recognised due to suboptimal myocardial definition on echocardiography and diverse phenotypic presentations.
Most commonly, non-compaction is associated with monogenic disorders, such as neuromuscular disorders, chromosomal abnormalities and congenital heart conditions. However, there are descriptions of acquired, reversible forms of LVNC found in sickle cell anaemia, athletes and pregnant women.13–15 The most common hypothesis posits that this acquired hypertrabeculation results as an adaptation to increased preload and myocardial wall stress in high cardiac output conditions. A prospective study of 102 primigravid pregnant women found that 26 subjects newly developed hypertrabeculation during pregnancy; hypertrabeculation subsequently significantly improved or resolved in all but two subjects during post-partum follow-up.14 Another study of 199 patients with heart failure with reduced ejection fraction found that 34.7% of subjects had prominent trabeculations, with 23.6% meeting formal criteria for LVNC.16 It is unlikely that this represents a large cohort of undiagnosed LVNC or an incomplete phenotype of this condition. It is more likely that the hypertrabeculation observed in this cohort was an adaptation to increased preload, as is seen in the previously discussed physiological states.
It is unclear whether our patient had two concurrent cardiomyopathies or whether the hypertrabeculation that developed was a result of his CS. Although evidence of concurrent non-compaction and sarcoidosis cardiomyopathy is unusual, one prior case report proposed that active inflammation from sarcoidosis in the trabeculation-rich myocardium may trigger the development of hypertrabeculation.1 Another hypothesis is that cardiac involvement of systemic sarcoidosis resulted in end-stage cardiomyopathy. As an adaptation to increased preload and worsening myocardial dysfunction, compensatory biventricular hypertrabeculation may have developed in our patient similar to the acquired LVNC seen in the cohort of heart failure patients.16 However, we cannot exclude the possibility of pre-existing non-compaction cardiomyopathy prior to the development of CS.
Conclusion
CS and LVNC are both under-recognised and may coexist in patients with advanced cardiomyopathy. It remains unclear whether these pathologies are mutually exclusive or whether one pathology may provoke the development of the other and this is worthy of further investigation.