










Heart failure (HF) with preserved ejection fraction (HFpEF) is a heterogeneous disorder developing from multiple aetiologies with overlapping pathophysiological mechanisms. Circulating biomarkers reflect cardiac as well as non-cardiac abnormalities, and their measurements often provide insights into pathophysiological processes associated with HF. The clinical uptake of biomarkers for diagnosing HFpEF has generally been poor, with only cardiac natriuretic peptides (NPs) having emerged as clinically relevant.1 Indeed, current European Society of Cardiology (ESC)/Heart Failure Association guidelines provide a class IB recommendation for NPs for diagnosis of suspected HF and NPs are a major criterion for establishing the diagnosis of HFpEF.1
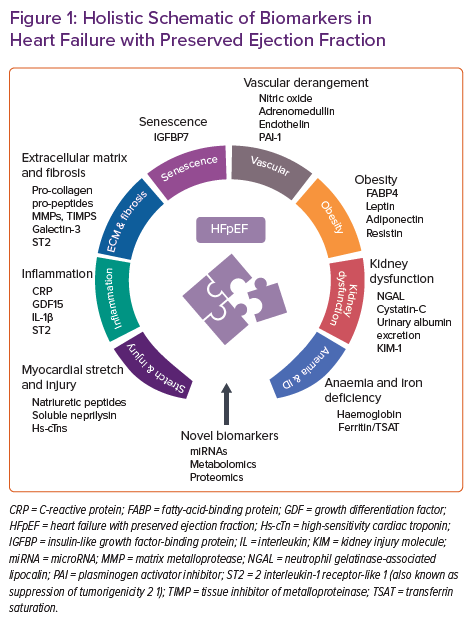
In this review we discuss identified HFpEF biomarkers derived from different pathological pathways. Most evidence is related to myocardial stretch and injury biomarkers, including NPs and cardiac troponins. Nevertheless, we also provide a comprehensive overview of biomarkers of inflammation, extracellular matrix derangements and fibrosis, senescence, vascular dysfunction, anaemia/iron deficiency and obesity. Finally, novel biomarkers from -omics technologies, including plasma metabolites and circulating microRNAs, are outlined briefly (Figure 1). A long road is ahead of us to better understand the complexity of HFpEF, but a comprehensive multimarker approach may be recommended to characterise specific HFpEF phenotypes/endotypes for optimising therapy and to stratify risk.
Myocardial Stretch and Injury
Natriuretic Peptides
Three endogenous NPs are secreted as pre-prohormones: atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP) and C-type natriuretic peptide. All these peptides act as hormones with pleiotropic effects contributing to cardiovascular homeostasis and pressure and volume overload counter-regulatory mechanisms.2 BNP and N-terminal proBNP (NT-proBNP) exhibit longer plasma half-lives (22 minutes and 70 minutes, respectively) compared to ANP (2 minutes); most of the evidence related to NPs in HF is from these two NPs, which are the gold standard diagnostic and prognostic biomarkers in HF patients.1 The pathophysiological mechanism of NP elevation in the setting of HF is well understood: BNP is produced primarily and secreted in the cardiac ventricles as a pro-hormone in response to myocardial stretch, to later be cleaved into vasoactive BNP and inactive NT-proBNP.3,4 These NPs have been directly correlated with several haemodynamic measures including left ventricular (LV) end-diastolic pressure, LV end-systolic and end-diastolic volumes and pulmonary capillary wedge pressure.5,6
The use of NPs has been rigorously studied as a diagnostic tool in HFpEF (Figure 2). A recent meta-analysis of 51 studies found that NPs have reasonable diagnostic performance in the detection of HFpEF in a non-acute setting (area under the receiver operating characteristics curve 0.80; 95% CI [0.73–0.87]; I2=86%) and are also a useful tool for ruling out diastolic dysfunction.7 Interestingly, lower levels of NT-proBNP are found in patients with HFpEF in comparison with those with HF with reduced ejection fraction (HFrEF),8,9 possibly because of the association of NPs with end-diastolic wall stress that appears to be lower in HFpEF.10 Obokata et al. attributed the lower NPs in HFpEF to lower wall stress due to pericardial restraint, which might be a dominant mechanism in some HFpEF patients.11 It is also remarkable that several cardiac and non-cardiac causes may impact NP levels, and they should be considered when attempting HFpEF diagnosis. Increased levels of NPs are evident in AF, the elderly and in people with severe renal impairment. In contrast, NP levels tend to be lower in obese patients, irrespective of volume status.5 These characteristics are frequently found in HFpEF patients, leading to evident variations in NP concentrations when different studies are compared. The observational study conducted by Anjan et al. found that nearly one-third of HFpEF patients had BNP levels below 100 pg/ml, while the I-PRESERVE trial revealed a median NT-proBNP concentration of 341 pg/ml and the observational study performed by Moliner et al. found a median NT-proBNP concentration of 956 pg/ml in HFpEF patients.8,12 Despite these considerations, the optimal cut-offs for distinguishing HFpEF and HFrEF from controls without HF appear to be remarkably similar, without differences according to HF phenotype in the diagnostic algorithm recommended in the ESC guidelines.1,13
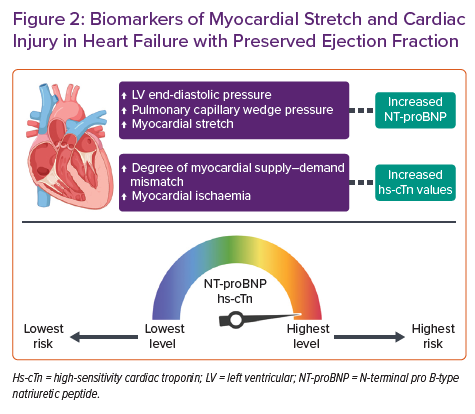
The utility of NPs in HFpEF is not restricted to their diagnostic capacity. Several studies have also investigated their ability to identify patients at risk for adverse events. A recent unsupervised cluster analysis based on a broad range of circulating biomarkers found that higher levels of NT-proBNP identify a subgroup of HFpEF patients (who also shared higher levels of cardiac troponin) who are at the highest risk of death and/or HF hospitalisation (62.8% over a median follow-up of 21 months).14
In the acute setting, NT-proBNP level is also considered a strong independent predictor of all-cause mortality, as recently described in the study by Lopuszynski et al. performed in a cohort of patients with chronic HF exacerbation and HFpEF.15 In this respect, previous studies have also reported equal prognostic significance of either admission levels or discharge levels of NPs in patients with HFpEF hospitalised for acute decompensated HF.16 NPs also confer the same relative risk information in HFpEF as in HFrEF in this acute setting, with similar adjusted prognostic relative risks in patients with both phenotypes for the relative changes in NT-proBNP levels during hospitalisation (Figure 2).17
Finally, in the field of patient management, it has been suggested that measuring NP concentrations should be useful in guiding therapy in the whole spectrum of HF, including the group of patients with HFpEF.18,19 However, current evidence does not support the routine measurement of BNP or NT-proBNP to guide titration of therapy. In this regard, the widespread strategy of using elevated plasma concentrations of NPs for patient selection in HFpEF trials has also been questioned, due to the lack of or lower benefit of irbesartan or aldosterone antagonists in patients with HFpEF and higher baseline NP values in the I-PRESERVE and TOPCAT trials, or the absence of sacubitril/valsartan treatment effect modification according to baseline NPs in the PARAGON-HF trial.12,20,21
Soluble Neprilysin
In the cardiovascular system, neprilysin cleaves numerous vasoactive peptides. Some of these peptides have vasodilating effects (including NPs, adrenomedullin and bradykinin), and others have vasoconstrictor effects (angiotensin I and II and endothelin [ET]-1, among others).22
Neprilysin serum levels (sNEP) exhibited significant prognostic value in both chronic and acutely decompensated HF.23–26 However, in patients with HFpEF results were controversial, perhaps due to different sNEP quantification methods.27,28 It is not only the prognostic role of sNEP that has been controversial: even blood sNEP concentrations can be very dissimilar with large differences among studies – some showing lower levels in HFpEF than in controls and others showing higher levels in HFpEF than in HFrEF patients.23,27,29 The correct quantification of sNEP remains a challenge that needs to be overcome to suppress potential biases regarding the interpretation of the different studies.30 Interestingly, some sNEP quantification methods showed that circulating sNEP was catalytically active.31
With the hypothesis that higher sNEP levels would correlate with lower NP levels, worse diastolic function, and subsequent clinical incident HFpEF, Reddy et al. performed a population study with 1,536 participants from Olmsted County, Minnesota.32 The authors found that low sNEP was paradoxically associated with worse diastolic dysfunction and hypertension but not with outcomes, including incident HF, over a median of 10.7 years of follow-up.
Due to these controversial results the assessment of sNEP in HFpEF patients is not currently recommended.30 However, it has recently been speculated that higher sNEP levels may identify HFpEF patients who might benefit from treatment with sacubitril/valsartan.33
High-sensitivity Cardiac Troponin
High-sensitivity cardiac troponin (hs-cTn) is universally recognised for its central role in defining myocardial injury in patients with acute coronary syndrome. However, hs-cTn can also predict the development of HF and reflect ongoing myocardial injury in the wide spectrum of HF.34–36 More specifically, elevated hs-cTn discriminates a subgroup of patients with HFpEF who have ongoing myocardial damage, higher wall stress or impaired microcirculation, as evidenced in a recent mechanistic study performed by Obokata et al.37 They assessed the relationship between troponin elevation and HFpEF physiology in 38 HFpEF patients and 20 control patients. HFpEF patients were found to have significantly higher troponin levels at rest, with the degree of elevation directly correlated to higher pulmonary capillary wedge pressure and worse systolic and diastolic tissue Doppler velocities. Troponin levels were also correlated with reductions in oxygen supply and a corresponding greater degree of supply–demand mismatch.
Hs-cTn has been shown to be of value in predicting the onset of HFpEF over a very long period in high-risk subjects in the general population and the elderly.38,39 The role of hs-cTn as a predictive biomarker in chronic HFpEF has also been a matter of research. Several studies have identified a significant association between elevated hs-TnT at admission and at discharge with adverse events in patients hospitalised with decompensated HFpEF.40–42 The predictive capabilities of hs-cTnT and hs-cTnI for secondary events in HFpEF have also been assessed. Both hs-cTnT and hs-cTnI are elevated in chronic HFpEF and are independently associated with poorer outcomes in men (HR 3.33; 95% CI [1.82–6.09]) than in women (HR 1.35; 95% [CI 0.94–1.93]). The predictive performance for composite outcomes was better for both hs-cTn assays in HFpEF than in HFrEF, but the strongest performance in HFpEF appeared to be from hs-TnT. The prognostic role for hs-cTn assays in HFpEF also has a sex-specific concern, as the more sensitive hs-cTnI assay appears to be a better predictor of outcome in men than in women (Figure 2).43
When combining the mechanistic data with the described associations of hs-cTn with adverse clinical outcomes, it appears reasonable to proclaim hs-cTn as a surrogate for a clinically meaningful HFpEF endpoint. This issue was partially addressed in the PARAGON-HF trial.44 The investigators found not only that hs-TnT was reduced by sacubitril/valsartan therapy compared with valsartan, but also that patients with a decrease in hs-TnT from randomisation to 16 weeks to a value to at or below the median value of 17 ng/l subsequently had lower risk of the composite outcome compared with those who had persistently elevated hs-TnT values.44 Further research will be required to determine whether therapies targeting the mechanism of hs-cTn elevation can improve symptom burden and clinical outcomes in the HFpEF population.
Inflammation
C-reactive Protein
C-reactive protein (CRP) may activate the complement system and stimulate cytokine production and thereby cause myocyte loss and promote LV remodelling and dysfunction.45 CRP has been shown to attenuate nitric oxide (NO) production and has a direct proinflammatory effect on human endothelial cells.46,47 Thus, CRP might worsen HF through multiple mechanisms. There is a close relationship between the number of comorbidities and plasma CRP level.48 Serum levels of CRP and pentraxin (PTX)-3, an acute phase protein of the PTX superfamily that also includes CRP, were both found to be significantly higher in HFpEF patients when compared with the non-HF reference group. PTX-3 was found to be an independent inflammatory marker correlated with the presence of LV diastolic dysfunction and HFpEF (Table 1).49
In the CANTOS trial, when canakinumab succeeded in lowering high-sensitivity CRP below 2 mg/l, the likelihood of HF hospitalisation was lower.50 This study did not discriminate between HFrEF and HFpEF, yet patients were old and had a high prevalence of obesity, diabetes and arterial hypertension.
Interleukin-1β
Interleukin (IL)-1β is a member of the IL 1 family of cytokines. This cytokine is produced by activated macrophages as a proprotein which is proteolytically processed to its active form by caspase 1 (CASP1/interleukin-1-converting enzyme). This cytokine is an important mediator of the inflammatory response and is involved in a variety of cellular activities, including cell proliferation, differentiation and apoptosis. Preclinical data show that the IL-1 family of cytokines contributes to cardiac dysfunction, and IL-1 blockade with anakinra (a recombinant IL-1 receptor antagonist) in patients with HF may improve cardiorespiratory fitness and prevent HF hospitalisations (Table 1).51 A randomised study in 31 patients with HFpEF (D-HART2) showed significant increases in treadmill exercise time, lower NT-proBNP levels and improved quality-of-life measures in the group treated with anakinra, without significant change in peak oxygen consumption.52
Interleukin 1 Receptor-like-1
Interleukin-1 receptor-like 1, also known as suppression of tumorigenicity 2 (ST2) has multiple isoforms, including a transmembrane form (ST2 ligand; ST2L) and a soluble circulating form (soluble ST2; sST2). Both ST2L and sST2 are expressed by cardiomyocytes and cardiac fibroblasts in response to mechanical stress, and both isoforms bind to IL-33. IL-33 is also induced by cellular stretch and apparently protects against fibrosis and hypertrophy in mechanically strained tissues via activation of myeloid differentiation primary response gene 88, interleukin-1 receptor-associated kinase, extracellular signal-regulated kinase and, ultimately, nuclear factor-κB.53
In HF, sST2 production by endothelium and the lungs is upregulated. In in vitro and in vivo models, ST2L transduces the effects of IL-33, while excess circulating sST2 leads to cardiac fibrosis and remodelling and ventricular dysfunction. It is proposed that circulating sST2 acts as a decoy receptor for IL-33, such that high levels of sST2 block the favourable effects of IL-33 by limiting activation of the cascade triggered by the IL-33/ST2L interaction. Thus, higher levels of sST2 are associated with increased myocardial fibrosis, adverse cardiac remodelling and worse cardiovascular outcomes (Table 1).54
In HFpEF, sST2 levels have been associated with proinflammatory comorbidities, right ventricular (RV) pressure overload and dysfunction, and systemic congestion, but not with LV geometry or function. These data suggest that sST2 may be a marker of systemic inflammation in HFpEF and potentially of extracardiac origin.55 Indeed, the Paulus and Tschöpe novel paradigm for HFpEF, of comorbidities driving myocardial dysfunction and remodelling through microvascular endothelial inflammation, has now been validated in several clinical cohorts and animal models.

Growth Differentiation Factor 15
Growth differentiation factor 15 (GDF15) is a member of the transforming growth factor β cytokine superfamily that is highly expressed in states of inflammatory stress.
GDF15 integrates information from cardiac and extracardiac disease pathways that are linked to the incidence, progression and prognosis of HF. Increased circulating levels of GDF15 are associated with an increased risk of developing HF in apparently healthy individuals (Table 1).56 After an acute coronary syndrome, elevated levels of GDF15 are indicative of an increased risk of developing adverse LV remodelling and HF. In patients with established HF, the levels of GDF15 and increases in GDF15 over time are associated with adverse outcomes. The information provided by GDF15 is independent of established risk factors and cardiac biomarkers, including BNP (Table 1).56
In clinical studies, median GDF15 values were similarly elevated in HFpEF and HFrEF, whereas NT-proBNP was significantly lower in HFpEF than in HFrEF. The similarly elevated levels and independent prognostic utilities of GDF15 in HFrEF and HFpEF suggest that beyond haemodynamic stress (NT-proBNP), inflammatory injury (GDF15) may play an important role in both HF syndromes.57
Extracellular Matrix and Fibrosis
Pro-collagen Propeptides
One of the hallmarks of interstitial fibrosis is deposition of types I and III fibrillar collagen. The collagen precursor, pro-collagen, consists of three polypeptide chains arranged in a triple helix, with non-helical N-terminal and C-terminal sequences. The N-terminal and C-terminal peptides are cleaved by endopeptidases after pro-collagen has been secreted from the cell.58
Elevated levels of pro-collagen type III N-terminal peptide (PIIINP) have been observed in individuals with hypertension, dilated cardiomyopathy, hypertrophic cardiomyopathy and recent MI, suggesting that the circulating peptide may be a useful marker of active myocardial collagen synthesis.59 In a retrospective analysis from RALES, high baseline levels of circulating PIIINP were predictive of death and hospitalisation in HF patients. Interestingly, the benefit of spironolactone, a putative anti-fibrotic agent, was observed primarily in participants with plasma PIIINP levels above the median (Table 1).60
Serum C-terminal propeptide of procollagen type I (PIP) strongly correlates with the turnover of extracellular cardiac matrix proteins and fibrosis. In this hypothesis-generating, mechanistic trial in stable HFpEF patients with type 2 diabetes (T2D), neither long-term administration of torasemide nor furosemide was associated with a significant effect on myocardial fibrosis, as assessed by serum PIP. Further studies are urgently needed in this field. More specific diuretic and anti-fibrotic treatment strategies in T2D and/or HFpEF are warranted.61
Matrix Metalloproteases and Tissue Inhibitor of Metalloproteinase
Matrix metalloproteases (MMPs) are calcium-dependent zinc-containing endopeptidases. These enzymes are capable of degrading all kinds of extracellular matrix proteins, but can also process a number of bioactive molecules. They are known to be involved in the cleavage of cell surface receptors, the release of apoptotic ligands (such as Fas ligand) and chemokine/cytokine inactivation. MMPs are also thought to play a major role in cell behaviours such as cell proliferation, migration, differentiation, angiogenesis, apoptosis and host defense.62 Tissue inhibitor of metalloproteinase (TIMP) is a natural glycoprotein inhibitor of the MMPs.
MMP2 and MMP9 activity and TIMP1 protein levels were enhanced in haemodynamic models of HFpEF, while metabolic models showed no changes in MMP2, -8, -9, -11, -14, -15, TIMP-1, -2 and -3 mRNA expression.63 In a PARAGON-HF collagen biomarker substudy, sacubitril/valsartan decreased TIMP1 by 8% compared with valsartan alone, consistently in men and women and patients with LV ejection fraction (LVEF) above or below the median of 57%. Higher levels of TIMP1 at baseline and increases at 16 weeks were associated with higher primary endpoint event rates (Table 1).64
Galectin-3
Galectin-3 is a β-galactoside-binding member of the lectin family and is encoded by a single gene, LGALSS3, located on chromosome 14. Galectin-3 is an ~30 kDa protein and contains a carbohydrate recognition binding domain of ~130 amino acids that enables the specific binding of β-galactosidases.
Galectin-3 has been linked to HF development and is implicated in a variety of processes that are thought to play an important role in the pathophysiology of HFpEF, such as myofibroblast proliferation, fibrogenesis, tissue repair, inflammation and ventricular remodelling.65,66 In experimental studies, several rodent models of pressure overload, such as hypertension and mice subjected to aortic constriction, have shown significant increases in myocardial, renal and vascular galectin-3 expression.65,67,68
Galectin-3 levels have been shown to correlate with HF severity and are highly associated with renal function.69–73 Changes in galectin-3 over time were shown to have an important prognostic value in HF patients, with increases in galectin-3 independently associated with an increased risk of all-cause mortality and HF hospitalisations.74–76 A ‘galectin-3-positive phenotype’ may predict the development of HFpEF in patients with co-morbidities and could be useful for phenotyping, risk stratification, and therapeutic targeting of those individuals where fibrosis is a major contributor to the syndrome (Table 1).77 In addition, galectin-3 is able to identify HF patients at low risk for adverse events.78
Vascular Mechanisms
Nitric Oxide
NO is produced by the enzymatic action of NO synthase (NOS) in vascular endothelium. Vascular actions of NO include direct vasodilation, indirect vasodilation by inhibiting vasoconstrictor influences, along with anti-thrombotic, anti-inflammatory and anti-proliferative effects.79 Low myocardial cyclic guanosine monophosphate (cGMP)-dependent protein kinase type I (PKG) activity is associated with raised cardiomyocyte resting tension and increased myocardial nitrosative/oxidative stress in HFpEF patients compared with HFrEF patients and patients with aortic stenosis.80 In several cohorts, HFpEF patients had lower serum NO-derived metabolites than those of HFrEF patients or controls without HF.81,82 However, this lower serum NO level did not correlate with the degree of LV hypertrophy or myocardial fibrosis by imaging.82 Thus, reduced NO bioavailability is considered to be an integral part of the inflammatory pathogenesis that results in myocardial dysfunction in HFpEF (Table 2).83 In a rat model of T2D, long-term application of the phosphodiesterase-5A inhibitor vardenafil, started in the prediabetic phase, effectively prevented development of HFpEF, reduced the pathophysiological features of T2D-associated cardiomyopathy and restored the activity of the cGMP–PKG axis.84 Unfortunately, human clinical trials testing NO as a therapeutic strategy have not demonstrated clinical benefits in HFpEF.85–88 Recently, results from a mouse model of HFpEF demonstrated that genetic inhibition of inducible NOS improves ventricular relaxation and exercise performance, providing a biological explanation for the failure of NO-inducing approaches as therapeutic strategies.89
Adrenomedullin
Adrenomedullin (ADM) is a potent vasodilator peptide, with additional immunomodulating, antiproliferative and antiapoptotic effects.90 ADM gene expression is significantly increased in the Dahl salt-sensitive rat model of HFpEF.91 In a large community-based study, mid-regional pro-ADM (MR-proADM) could help in identifying new-onset HFpEF.92 In the same way, in HFpEF patients from the KaRen biomarker substudy, ADM predicted HF severity and prognosis.38 In a cross-sectional analysis from the prospective cohort program DIAST-CHF, MR-proADM was independently associated with submaximal exercise capacity when added to the base model, which included classical risk factors for HFpEF.93 In a prospective study, HFpEF patients displayed higher levels of MR-proADM at rest and during exercise in comparison with subjects without HF. These high neurohormone levels were associated with pulmonary haemodynamic derangements, limitations in RV functional reserve, reduced cardiac output and more profoundly impaired exercise capacity in HFpEF (Table 2).94 Recently, ADM has been identified as an important marker of volume expansion and prognostically relevant feature in HFpEF patients with an obese phenotype with important prognostic implications.95
Endothelin
ET is the most potent vasoconstrictor peptide known in humans. It is produced in higher concentrations by endothelial cells and is involved in the regulation of vascular tone. In the LURIC study, a single cut-off point (0.8 fmol/ml) of Big-ET-1 was significantly associated with higher incidence rates of cardiovascular death at both intermediate and high NT-proBNP levels.96 In a post hoc analysis of the RELAX trial, plasma ET-1 levels were significantly associated with lower exercise oxygen consumption both at baseline and longitudinally over 24 weeks.97 ET-1 levels were predictive of 1-year HF hospitalisation and were associated with long-term mortality in HFpEF patients (Table 2), suggesting that ET-1 could be a biomarker in HFpEF and a potential therapeutic target.98
Following this, in a prospective study with HFpEF patients, the dual ET-A/ET-B receptor inhibitor macitentan improved HFpEF by abrogating adverse cardiac remodelling via antihypertrophic mechanisms and by reducing stiffness.99 In the same population, sitaxsentan, a selective ET-A receptor antagonist, increased exercise tolerance but did not improve any of the secondary endpoints such as LV mass or diastolic function.100
Plasminogen Activator Inhibitor-1
Plasminogen activator inhibitor-1 (PAI-1) is a serine protease inhibitor that functions as the principal inhibitor of tissue plasminogen activator and urokinase, the activators of plasminogen and hence fibrinolysis. Thus, elevated PAI-1 is a risk factor for thrombosis and atherosclerosis.101 Accordingly, PAI-1 levels were increased in patients with HFpEF in comparison with healthy controls, as were D-dimer and tissue plasminogen activator levels, suggesting that HFpEF is associated with a procoagulant state.101 The tissue plasminogen activator/PAI-1 complex was an independent predictor of mortality from all causes and from cardiovascular causes in patients with HFpEF from the LURIC study (Table 2).102 Nevertheless, in a large study that included four longitudinal community-based cohorts, only NPs and the urinary albumin-to-creatinine ratio were associated with HFpEF,103 while PAI-1 showed only a suggestive association.104

Senescence
Insulin-like Growth Factor-binding Protein 7
The major function of insulin-like growth factor-binding protein (IGFBP) 7 is to regulate the availability of insulin-like growth factors. It also stimulates cell adhesion and is associated with inflammation, cellular senescence, tissue aging and obesity. Some proteins involved in inflammation, such IGFBP7, formed a conserved network in HFpEF across two independent cohorts from the PROMIS-HFpEF study and may mediate the association between comorbidity burden and echocardiographic indicators of worse haemodynamics and RV dysfunction.105 Patients with a pan-inflammatory phenotype exhibited the highest circulating levels of inflammatory mediators, more comorbidity, more HF hospitalisations, higher left-atrial volume index and NT-proBNP level, worse renal function, the highest levels of fibrotic biomarkers such IGFBP7 and the lowest functional capacity, in an HFpEF study that used unsupervised machine learning.106 Nevertheless, higher concentrations of IGFBP7 were associated with increased risk of cardiovascular events in patients from the I-PRESERVE trial, but after multivariable adjustment this association was no longer present.107
Regarding diastolic function, in a cohort of HFpEF patients IGFBP7 was elevated and associated with markers of diastolic dysfunction, HF severity and prognosis (Table 2).108 In HFpEF patients randomised to receive sacubitril/valsartan versus valsartan, higher concentrations of IGFBP7 were associated with abnormalities in diastolic filling and left atrial dilation. Treatment with sacubitril/valsartan resulted in lower IGFBP7 concentrations than did treatment with valsartan.109 In the same way, in HFpEF patients from the RELAX trial, higher baseline IGFBP7 was modestly correlated with worse diastolic function but its change at 24 weeks was significantly correlated with changes in diastolic function and exercise capacity.110 Another study, comparing controls with normal diastolic function and asymptomatic LV diastolic dysfunction with HFpEF patients, showed a rise in IGFBP7 levels in HFpEF patients that ran parallel to worsening diastolic function.111
Obesity
Fatty-acid-binding Protein 4
Fatty-acid-binding proteins (FABPs) are intracellular lipid chaperones.112 FABP4 (also known as adipocyte FABP or aP2), plays an important role in the development of obesity, insulin resistance, diabetes and atherosclerosis.113 Moreover, FABP4 is associated with cardiac remodelling and both left and right ventricular dysfunction.114
FABP4 is highly expressed in response to increased lipolytic signals of catecholamine and NPs. FABP4-deficient mice develop dietary obesity due to reduced efficiency of lipolysis, but not insulin resistance or diabetes.115 It has been postulated that FABP4 activates hormone-sensitive lipase in adipocytes.
A biomarker study of TOPCAT has shown an association between FABP4 and incident risk of death or HF admission, independent of the Meta-Analysis Global Group in Chronic Heart Failure risk score (Table 3);116 similar results were found in another cohort when adjusted for age, sex and NT-proBNP level. Recently, Harada et al. reported that event-free survival is significantly decreased in patients with HFpEF and FABP4 levels ≥43.5 ng/ml.114
Leptin
Leptin is an adipocyte-derived circulating protein that modulates food intake and body weight, suggesting that it signals to the brain the magnitude of fat stores. However, in several rodent models of obesity, leptin levels are increased, suggesting resistance to suppress satiety.
In mouse models of obesity with high leptin concentrations, this protein may be protective against LV hypertrophy.117 Obese diabetic dB/dB mice (with functionally inactive leptin) show diastolic dysfunction with preserved LVEF, associated with cardiomyocyte hypertrophy, fibrosis and microvascular rarefaction (Table 3).118 Leptin also acts on the kidney to promote sodium retention, thus increasing the secretion of aldosterone, the activity of the renal sympathetic nerves and the Na+-K+ ATPase in the renal tubules119 (opposite effect of sodium–glucose cotransporter 2 inhibitors) (Table 3). Patients with HFpEF have a particularly elevated level of leptin, which is associated with better outcome in HFrEF but not in HFpEF120 (neither on decompensated HF admission nor in New York Heart Failure Association [NYHA] class).92
Adiponectin
Adiponectin is an adipokine implicated in energy metabolism, increasing insulin sensitivity and beta-oxidation and reducing gluconeogenesis (Table 3).121 In chronic HF patients, adiponectin is considered to be a marker of catabolic state and disease progression, and low body fat is associated with higher levels of adiponectin.122 Beyond metabolic control, adiponectin has been linked to a cardiovascular risk profile (insulin resistance, arteriosclerosis and inflammation).123
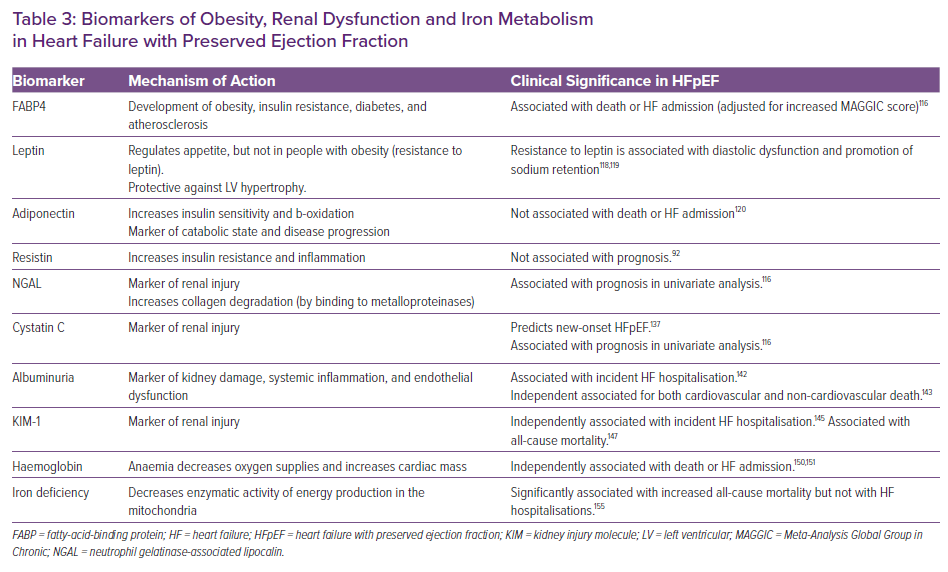
In experimental models, adiponectin deficiency leads to hypertension, LV hypertrophy and diastolic dysfunction. Accordingly, overexpression of adiponectin in aldosterone-infused mice ameliorated cardiac remodelling without affecting blood pressure and LVEF.124 Adiponectin levels are raised in both HFpEF and HFrEF. Patients with plasma adiponectin above a mean of 13.5 mg/l had a 3.4-fold increase in mortality risk, but higher levels have been associated with adverse outcome and poor prognosis only in HFrEF.120,125
Resistin
Resistin is a polypeptide, initially discovered in adipocytes, that has been associated with both insulin resistance and inflammatory response.126 Administration of anti-resistin antibodies improves blood sugar in mice with diet-induced obesity.127 Moreover, in collared rabbit carotid arteries, resistin has been shown to aggravate atherosclerosis by stimulating monocytes, endothelial cells and vascular smooth muscle cells.128 Resistin has been linked with incident HF, and plasma levels are significantly elevated in HF.126,129,130 Nevertheless, in HFpEF patients, resistin has not predicted outcomes in univariable analysis (Table 3).97
Kidney Dysfunction
Neutrophil Gelatinase-associated Lipocalin
Neutrophil gelatinase-associated lipocalin (NGAL or lipocalin-2) is a marker of early renal dysfunction.131 NGAL is massively upregulated after renal tubular injury, but also during inflammation and renal and myocardial ischaemia or infection.132 In addition, NGAL may also play a role in atherosclerosis and matrix degradation.
NGAL has been associated with a probable renal protective effect, since it can form complexes with siderophores, chelate the iron released by damaged tubules and prevent hydroxyl radical creation. Moreover, NGAL binds to the MMP-9, slows its degradation, and enhances collagen degradation.133 Plasma NGAL at discharge predicted 30-day outcomes in patients admitted for acute HF even more strongly than did NT-proBNP.134 In the TOPCAT biomarker sub-study, NGAL level tended to be associated with death or HF admission but this association lost significance upon multiple comparison (Table 3).116
Cystatin-C
Cystatin C (CysC) is secreted by nucleated cells at a constant rate, filtered and reabsorbed by the glomeruli, and then completely decomposed by intact renal tubules.134 CysC provides a more sensitive and accurate method for estimated glomerular filtration rate (eGFR) measurement: serum CysC reflects the GFR, while urine CysC reflects the degree of renal tubular damage. In addition, excess CysC may promote myocardial fibrosis and ventricular hypertrophy and increase atrial volume.135,136
CysC is a strong risk factor for new onset HFpEF (Table 3),137 and it is significantly associated with the NYHA classification, even after adjustment for eGFR.138 Besides, CysC is a strong and independent predictor of an unfavourable outcome on admission in HFpEF.139 In chronic HFpEF, CysC tended to be associated with death or HF admission, but without meeting significance in the multivariable analysis (Table 3).116
Urinary Albumin Excretion
Increased urinary albumin excretion (UAE) is considered to be an early marker of kidney damage. Albuminuria is considered to be a biomarker of systemic inflammation and endothelial and microvascular dysfunction.140 Moreover, albuminuria is associated with increased RV and LV remodelling and longitudinal systolic dysfunction.141
Urinary albumin-to-creatinine ratio >300 mg/g has been associated with a 4- to 5-fold increased risk of incident HF hospitalisation (Table 3).142 In established HFpEF, UAE is a powerful and independent predictor of both cardiovascular and non-cardiovascular death (Table 3),143 in each stratum of eGFR.141
Kidney Injury Molecule-1
Kidney injury molecule-1 (KIM-1), is a transmembrane glycoprotein expressed to regulate the regeneration and repair of the kidney after tubulo-interstitial damage.144 The urinary level of KIM-1 has been suggested as a clinically relevant biomarker of acute tubular injury. Higher urinary levels of KIM-1 have been associated with an increased risk of incident HF hospitalisations, even after adjustments for age, established risk factors, albuminuria and eGFR (Table 3).145 In HFrEF, KIM-1 has shown an association with prognosis; recent data also support the prognostic role of KIM-1 in patients with HFrEF on all-cause mortality (Table 3). 146,147
Anaemia and Iron Deficiency
Haemoglobin (Anaemia)
Anaemia modifies myocardial energy efficiency by lowering oxygen delivery to the myocytes and by increasing cardiac mass, LV end-diastolic pressure and ventricular remodelling. Haemoglobin level is inversely related to LVEF, and an increase in haemoglobin over time is associated with a decrease in LVEF.148
Defective iron usage, inappropriate erythropoietin production, and depressed bone marrow function due to renal dysfunction and neurohormonal and proinflammatory cytokine activation leading to increased myocardial workload are suggested causes of anaemia in HF.149
In a post-hoc analysis of the TOPCAT-Americas population, the risk of mortality and HF-hospitalisations increased significantly with a decrease in haemoglobin after extensive adjustment; similar data were found in a post-hoc analysis of the CHARM-Preserved trial (Table 3).150,151 Whether anaemia is a mediator or a marker of poor outcomes and HF severity is not clear, since correcting anaemia does not improve prognosis in HFrEF and no data have been published in HFpEF.
Iron Deficiency (Transferrin Saturation Index, Serum Ferritin)
Iron deficiency is defined as ferritin <100 or 100–299 µg/l with transferrin saturation <20%. Iron deficiency plays an important role in oxygen uptake, transport and storage; the cellular immune response; oxidative metabolism; and cellular energetics.152 At the cellular level, it is thought to decrease the enzymatic activity of both the Krebs cycle and the mitochondrial respiratory chain.153
Iron deficiency has been related to decreased exercise capacity (measured with both peak VO2 max and 6-minute walking distance) and quality of life in HFpEF patients.154 Moreover, a recent study has shown that iron deficiency in HFpEF is significantly associated with a 3.5-fold increase in all-cause mortality, but not with HF hospitalisations (Table 3).155 The on-going FAIR-HFpEF study is evaluating the effects of intravenous iron therapy on exercise capacity, mortality and HF-related hospitalisation rates, as has been previously reported in HFrEF patients.156
Novel Biomarkers
Circulating microRNAs
Circulating microRNAs (miRNAs) offer attractive potential as epigenetic disease biomarkers by virtue of their biological stability and ready accessibility in liquid biopsies. Numerous clinical cohort studies have revealed unique miRNA profiles in different disease settings, suggesting their utility as markers with diagnostic and prognostic applications.157
The discovery of microRNA clusters that are differentially expressed in HFrEF and HFpEF patients provides an approach to delineate the different pathobiological pathways underlying HFrEF and HFpEF. A putative biological pathway affected by a panel of eight HFpEF-related miRNAs (hsa-miR-193a-5p, hsa-miR-30a-5p, hsa-miR-106a-5p, hsa-miR-191-5p, hsa-miR-486-5p, hsa-miR-181a-2-3p, hsa-miR-660-5p and hsa-miR-199b-5p) has been reported as valuable in identifying HFpEF.158
However, there is no consensus on the choice of specific circulating miRNAs that might better serve as HFpEF biomarkers. A recent report found circulating miR-181c as a marker of the response to exercise training in patients with HFpEF.159 Further research is needed to understand their added value in diagnosis and prognosis at the clinic, beyond their research interest.
Metabolomics
Patients with new-onset HFpEF compared with patients with new-onset HFrEF display a different metabolic profile associated with comorbidities such as diabetes and kidney dysfunction. In an exploratory study, new-onset HFpEF patients had a diverging metabolite pattern compared with that of HFrEF patients, reflecting potential differences in pathophysiological mechanisms.160 First, HFpEF patients displayed elevated hydroxyproline reflecting fibrosis, elevated symmetrical dimethylarginine indicating oxidative stress, and elevated alanine, cystine and kynurenine reflecting a state of increased inflammation compared with HFrEF patients. Second, HFpEF patients had lower levels of cGMP and cyclic adenosine monophosphate, suggesting impaired cell signalling; lower L-carnitine, indicating mitochondrial dysfunction; and lower levels of lysoPC (18:2) associated with impaired lipid metabolism. Third, serine and arginine levels were lower in HFpEF than in HFrEF, reflecting endothelial dysfunction in HFpEF.160
Proteomics
Exploration of 92 proteins from the Olink cardiovascular II panel and their association with obese HFpEF has been recently reported in the LIFE-Heart study (999 patients with HFpEF and 999 patients without HF). Obese HFpEF patients exhibited higher circulating biomarkers of volume expansion (adrenomedullin), myocardial fibrosis (thrombospondin-2) and systemic inflammation (galectin-9, CD4) compared to obese non-HFpEF or lean HFpEF patients.
In the setting of HFpEF and diabetes, Hanff et al., using SomaScan assays and proteomics analysis of plasma from participants in the TOPCAT trial and the Penn Heart Failure Study, identified 10 proteins with significantly different expression in patients with HFpEF and diabetes.161 These proteins included fatty acid-binding protein, alpha-1-microglobulin/bikunin precursor, trafficking protein particle complex subunit 3, pigment epithelium-derived factor, tumour necrosis factor ligand superfamily member 15, ubiquitin-conjugating enzyme E2 G2, reticulon-4 receptor, insulin, cartilage intermediate layer protein 2 and apolipoprotein M. Of these, apolipoprotein M was found to mediate 72% of the association between diabetes and the risk of cardiovascular death, aborted cardiac arrest and HF hospitalisation.161
The use of SomaScan technology showed that patients with HFrEF, HF with mid-range ejection fraction (HFmrEF), and HFpEF had unique variations in circulating proteins which reflected distinct biological pathophysiologies. Bioinformatics analysis revealed biological themes that were unique to HFrEF, HFpEF and HFmrEF patients, suggesting that it may be possible to use proteomics assays to more accurately predict clinical phenotypes of HF patients.162 Further research is needed to validate these results and translate the proteomic data to the bedside.
The use of machine-learning algorithms applied to a wide range of biomarkers in HFpEF cohorts has identified several clusters a with different cardiovascular phenotypes and outcomes.163,164
Conclusion
The pathophysiological basis for identifying and classifying HFpEF based on a multimarker strategy seems logical and deserves further research. A cardiac-centred approach to HFpEF diagnosis using NPs is a good starting point, and is actually promoted by the Heart Failure Association through the ‘Peptide for Life’ initiative.165 However, a holistic approach including biomarkers that provide information on non-cardiac components of the HFpEF syndrome may enrich our understanding of the disease and may be useful in classifying HFpEF phenotypes/endotypes that may guide patient selection in HFpEF trials (Figure 1).166 Because of the risk of bias in current HFpEF biomarker studies, methodologically well-designed studies with a uniform reference diagnosis are urgently needed to determine the incremental value of circulating biomarkers for the diagnosis of HFpEF.167