








Heart failure (HF) affects 1–2 % of the population in developed countries and absorbs a significant amount of human and economic resources.1–3 It is a complex syndrome, characterised by a spectrum of symptoms and signs ranging from minimal loss of normal functional capacity to more severe symptoms refractory to medical therapy. It may be associated with different aetiologies and varying degrees of systolic and/or diastolic cardiac impairment.
Recently, understanding of the complex metabolic processes associated with the development of HF has been growing.4 As a consequence, HF is now understood to be a systemic, multi-organ syndrome with metabolic failure the basic mechanism. The failing heart may be defined as ‘an engine out of fuel’.5 Metabolic derangement in HF is not limited to the myocardium but extends to the skeletal muscles and contributes to the deterioration of exercise capacity in these patients, who can experience muscle weakness, fatigue, exercise limitation and dyspnoea, and to disease progression.4
This review will examine the role of metabolic abnormalities in HF and their therapeutic implications, with a focus on the only drug currently available worldwide.
Metabolic Processes in the Normal and Failing Heart
At rest, free fatty acid oxidation is the major source of energy for the myocardium. Up to 80 % of high-energy phosphates at rest are produced by the oxidation of free fatty acids. Glucose metabolism provides the remaining quantity of energy. The heart stores glucose as glycogen to be used during increased metabolic demands since glucose utilisation is 20–30 % more metabolic efficient than free fatty acid oxidation in producing high-energy phosphates per mole O2.6
At rest, the myocardium uses 15–20 % of its maximal oxidative capacity and adapts substrate utilisation during increased demand.6 A net increase in glucose and lactate uptake and utilisation without change in free fatty acid metabolism has been demonstrated during low to moderate intensity exercise.7–9 However, when the metabolic requirements of the myocardium exceed the limits of its metabolic reserve, an aerobic limit is reached. This threshold is higher when glucose is used as substrate rather than fatty acids.
Maladaptive energetics play an important role in the pathophysiology of the failing heart. HF is a ketosis-prone state as blood ketone bodies are increased in this syndrome. Blood ketone bodies and free fatty acid levels are higher during fasting and remain higher after glucose infusion in patients with chronic HF than controls.10 The blood levels of blood ketone bodies are related to the severity of cardiac dysfunction and neurohormonal activation in HF.11
A possible cause for these metabolic derangements in HF is myocardial insulin resistance that develops early in HF which limits the utilisation of glucose and favours the increased utilisation of free fatty acids for ketogenesis. All these changes lead to a reduction in the production of high-energy phosphates and therefore to a metabolically inefficient heart.
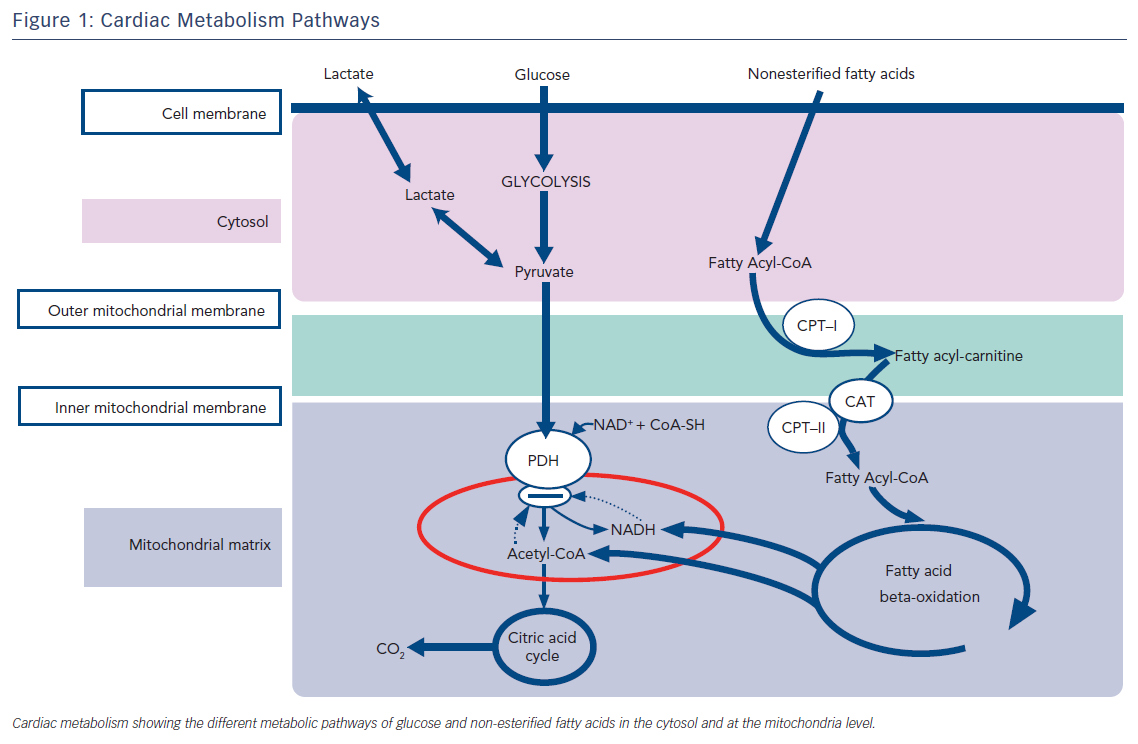
Therefore, in HF metabolic changes add to the main causes of the disease favouring its progression and reducing functional capacity. In some conditions, such as diabetes, metabolic derangements leading to inefficient production of high-energy phosphates may constitute the main cause of the disease and may also be responsible for the altered diastolic function observed in most patients with HF with preserved ejection fraction.
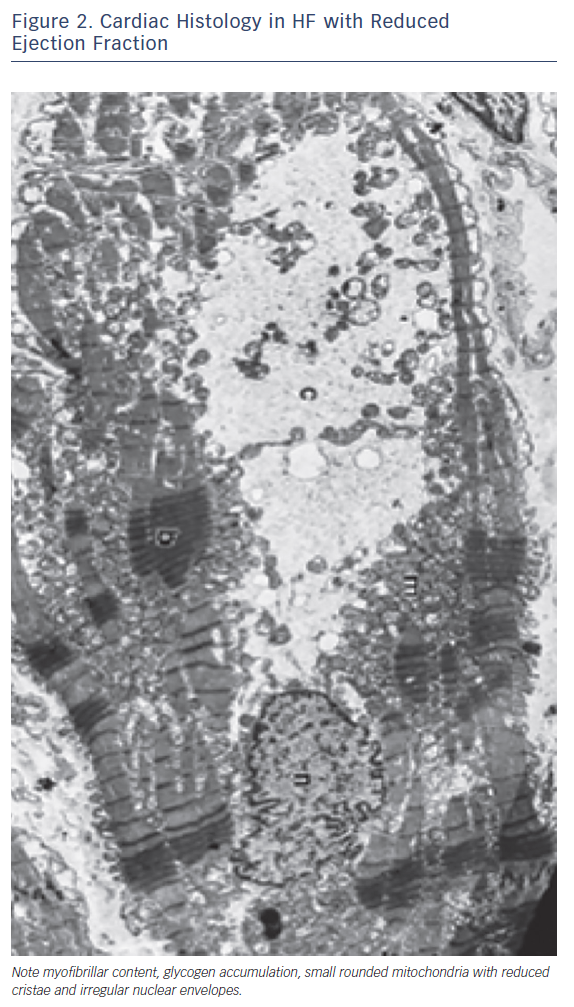
Other metabolic abnormalities, ranging from testosterone deficiency to a metabolic shift favouring catabolism and impairment in skeletal muscle mass and function, occur in patients with HF and may heighten the metabolic changes occurring in the glycolytic pathway.12 Changes in glucose utilisation lead to a deficiency in high-energy phosphate availability and reserve, and impaired contractility and relaxation.13 Furthermore, other processes, such as structural remodelling and oxidative stress, are also activated as a consequence of the metabolic derangements. All these metabolic alterations are defined as ‘metabolic remodelling’, i.e. remodelling of cardiac energy metabolism, which causes a decrease in energy production and a switch in energy substrate use. Therefore, metabolic remodelling contributes to the progression of HF and to it becoming worse causing progressive loss of myofibrillar content and shape and size of mithocondria (Figure 1).14,15 The reduction in skeletal and cardiac muscle production of high-energy phosphates leads to a progressive decline in both diastolic and systolic function and to the progression of left ventricular remodelling in a metabolic vicious circle.14 The metabolic changes induce maladaptive cellular changes, with decreases in mitochondrial cristae, cellular size, myofibrillar content and actino-myosin coupling. Therefore, interventions aimed at improving cardiac and skeletal muscle metabolism by optimising cardiac metabolism and improving high-energy phosphate production may be a complementary effective approach to the treatment of HF.16
Treatment of HF with Metabolic Agents
Neurohormonal antagonists, such as angiotensin-converting enzyme (ACE) inhibitors, beta-blockers or mineralocorticoid receptor antagonists, should be used to modify the progression of systolic dysfunction and to improve the prognosis of patients with chronic HF.17 Diuretics are used in combination with these drugs to relieve symptoms and signs of congestion. However, despite advances in pharmacotherapy for HF with newer drugs such as ivabradine and sacubitril/valsartan, the prognosis of patients with HF remains poor. Cardiac inotropes that increase cardiac energy consumption have been found to have negative effects on long-term prognostis. These effects may be related to the exhaustion of high-energy phosphates leading to an insufficient handling of Ca2+ favouring arrhythmias.
The modulation of cardiac metabolism with drugs to promote the preferential use of glucose and non-free fatty acid substrates by the mitochondria to increase metabolic efficiency and function of the failing heart was proposed in the late 1990s. However, only recently has enough clinical evidence accumulated to support the rationale and use of this.6
Several agents able to interfere with cardiac metabolism have been proposed in previous decades but, among all the proposed agents, only trimetazidine and perhexiline have been approved for human use. These two drugs, initially approved for the treatment of angina pectoris and myocardial ischaemia, both directly inhibit myocardial fatty acid oxidation and improve regional and global myocardial function.18 However, while trimetazidine is available worldwide (except in the US where its dossier has never been submitted), carnitine palmitoyltransferase (CPT) inhibitor perhexiline is not available in most countries amid doubts over its safety profile, although there is growing evidence of its efficacy in HF.19,20 Both drugs directly inhibit FFA oxidation by blocking 3-ketoacyl-coenzyme A thiolase (3-KAT), which shifts energy production from FFA to glucose oxidation. Several studies have shown the benefits of these compounds on myocardial function in patients with ischaemic and non-ischaemic HF.16
Trimetazidine has been more extensively studied in HF. The beneficial effects of trimetazidine in HF are related to improved of phosphocreatine (PCr) and ATP intracellular levels. This is of particular relevance as PCr/ATP levels are a significant predictor of mortality in HF.16 Indeed, trimetazidine has been shown to improve prognosis in patients with HF and reduced ejection fraction in a multicentre retrospective cohort study and in four meta-analyses.21 On this basis, its use in patients with HF has been advocated by many authors and it is supported by current guidelines.16
Efficacy of Metabolic Modulation in HF
The beneficial effect of metabolic modulation of cardiac metabolism in HF has been attributed to the shift of energy production from free fatty acid oxidation to glucose oxidation, which leads to increased production of high-energy phosphates and therefore to greater cardiac and skeletal muscle efficiency.22 The metabolic modulation through inhibition of free fatty acid metabolism is associated with an improvement of the cardiac phosphocreatine:ATP ratio by 33 %, which translates into a parallel increase of left ventricular function. This effect is associated with a reduction in the whole-body rate of energy expenditure, which suggests that the effect may be mediated by decreased metabolic demands in peripheral tissues. The decreased peripheral metabolic demands are the consequence of a greater muscular metabolic efficiency leading to reduced oxygen consumption per any given level of exercise. It has been shown that free fatty acid inhibition improves functional capacity and muscular strength in patients with HF, suggesting an effect that goes beyond the modulation of cardiac metabolism.23–32
Several clinical trials have demonstrated that metabolic modulation through inhibition of free fatty acid metabolism improves New York Heart Association (NYHA) HF class, exercise tolerance, quality of life, left ventricular ejection fracture and cardiac volumes in patients with ischaemic and non-ischaemic HF.23–34
More recently, studies have shown that optimising cardiac metabolism with trimetazidine may improve the prognosis of patients with HF. Randomised clinical trials demonstrated the effect of metabolic modulation of cardiac metabolism added to conventional therapy in improving functional class, left ventricular end-systolic volume and ejection fraction in patients with HF of various origins. An international multicentre cohort study on 669 patients with chronic HF21 has shown that the adjunct of trimetazidine to conventional therapy is effective in reducing mortality and hospitalisations, and improves long-term survival in people with HF. However, further prospective trials that are adequately powered to specifically look at mortality are needed. Four meta-analyses of the available randomised controlled trials have consistently shown that trimetazidine ameliorates cardiac function for ischaemic and non-ischaemic HF, reduces mortality, cardiovascular events and hospitalisation.35–41
The first meta-analysis pooled data from 17 RCTs, which included 955 patients with HF and found that treatment with trimetazidine is associated with NYHA class reduction, increased exercise tolerance and improvement of LVEF in patients with HF of both ischaemic and non-ischaemic aetiology. The analysis found that the use of trimetazidine reduces rates of cardiovascular events and hospitalisations as well as all-cause mortality in patients with HF.
In a second meta-analysis performed by Zhang et al., based on data from 16 RCTs with 884 patients with HF, trimetazidine was shown to improve NYHA class, increase exercise tolerance, improve LVEF and decrease left ventricular end-systolic and end-diastolic diameters; trimetazidine also reduced rates of hospitalisation but not all-cause mortality. An updated meta-analysis by Zhou and Chen that included data from 994 patients with HF from 19 RCTs confirmed the improvement in NYHA class, cardiac volumes and LVEF in patients treated with trimetazidine. This analysis confirmed the reduction in the rate of hospitalisations for HF.
More recently, Grajek and Michalak conducted a meta-analysis assessing the effect of trimetazidine on all-cause mortality rates in patients with HF. They included 326 patients from three RCTs who received trimetazidine in addition to standard pharmacological therapy for HF and found a significant reduction in all-cause mortality rate among patients receiving trimetazidine.
Because of the clinical evidence on its efficacy, trimetazidine has been included in the ESC/HFA 2016 guidelines for the treatment of HF with reduced ejection fraction for patients with HF of ischaemic aetiology; it is used as add-on to an ACE inhibitor (or an angiotensin receptor blocker if ACE inhibitors are not tolerated), a beta-blocker and a mineralocorticoid receptor antagonist.
Finally, new metabolic agents are emerging as promising therapeutic candidates for HF, as reviewed in detail elsewhere.42 Briefly, there is evidence on efficacy and good tolerability of istaroxime, a drug with lusitropic and inotropic digoxin-like properties, in a phase II clinical trial (HORIZON–HF)43 on patients with HF and reduced ejection fraction. Another compound, omecamtiv mecarbil, a direct activator of myocardial myosin ATPase, has been shown in a clinical trial (ATOMIC–AHF)44 to increase systolic ejection time with good safety; it is being tested in an international multicentre phase III clinical study to test its efficacy in reducing cardiovascular events.
Conclusions
HF is associated with a maladaptive cardiac metabolism that impairs cardiac function. Understanding the role of cardiac metabolism in HF will open new therapeutic avenues for patients.39-40 Modulation of cardiac metabolism may be especially beneficial in patients with altered glucose metabolism, where the systemic metabolic derangements have profound effect on myocardial function, increasing the risk of HF and its mortality.41
Clinical Perspective
- Heart failure is associated with maladaptive cardiac metabolism.
- Cardiac metabolism in heart failure is shifted towards the use of less energy-efficient free fatty acids.
- Improvement of glucose utilisation by the cardiac cells is associated with improved cardiac function.
- Modulation of cardiac metabolism improves left ventricular function and exercise capacity in heart failure.