










Coronavirus disease 2019 (COVID-19) has evolved into a global pandemic, having affected more than 2.3 million people and claiming more than 160,000 lives. Infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus predominantly causes fever (77–98% of cases), fatigue (52–75%) and cough (60–81%).1,2 While it primarily affects the respiratory system, it also appears to have a unique interplay with the cardiovascular system.
Patients with pre-existing cardiovascular comorbidities appear to be at the highest risk for mortality from COVID-19, along with the elderly. The disease also contributes to cardiovascular complications, including acute coronary syndromes, arrhythmias, myocarditis, acute heart failure and, in the most severe cases, cardiogenic shock and death.3
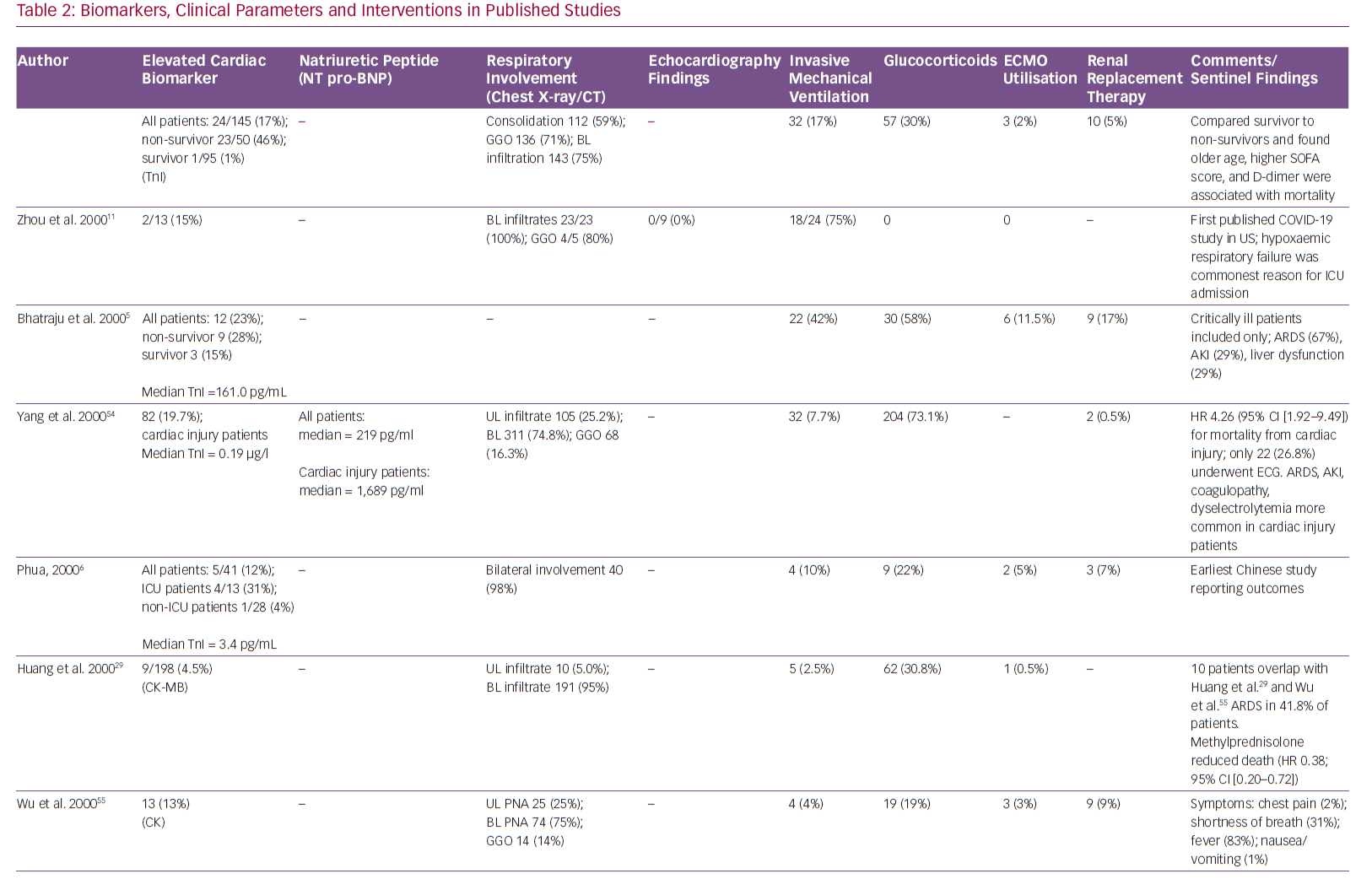
Although only a few population studies have detailed the spectrum of cardiovascular complications, the high prevalence of myocardial injury in patients with COVID-19 is suggested by frequently elevated cardiac biomarkers. Elevated troponin levels are noted in 7–28% of COVID-19 patients on presentation, some associated with depressed left ventricular function and haemodynamic shock.4–7 Although an elevation in cardiac troponin is a sensitive marker for myocardial injury, it does not distinguish between the various aetiologies of injury. Multiple potential mechanisms of acute myocardial injury from the viral infection have been proposed.8
The purpose of this article is primarily to summarise the available literature (Tables 1 and 2) on various proposed mechanisms of myocardial injury related to COVID-19 (Figure 1).
Acute Coronary Syndromes
Although the reported incidence of acute coronary syndrome (ACS) has gone down during the COVID-19 pandemic, it is possible this is because patients are being hesitant in visiting hospitals for their medical care.9 However, a variety of syndromes mimicking ACS have been reported in people with COVID-19 infection.
In a case series of 18 patients with COVID-19, 56% were found to have ST-elevation at time of presentation, with the rest developing it at some point during hospitalisation.10 Focal ST elevations were present in 78% of patients. A total of nine patients (50%) underwent coronary angiography but only six patients (33%) were eventually found to have obstructive coronary artery disease (CAD). Inferior and lateral ST segment elevations were most common, and ECG findings were more likely to be focal in patients with obstructive CAD. Of note, all 18 patients had elevated D-dimer levels.
In a recently published retrospective study of 191 COVID-19 patients from two separate hospitals in China, the incidence of elevation in high-sensitivity cardiac troponin I (cTnI; >28 pg/ml) was 17%, and it was significantly higher among non-survivors (46% versus 1%, p<0.001).11 Furthermore, elevation of this biomarker was noted to be a predictor of in-hospital death (univariable OR 80.07, 95% CI [10.34–620.36], p<0.0001). The most abrupt increase in cardiac troponin I in non-survivors was noted beyond day 16 after the onset of disease.
In the same study, the incidence of acute cardiac injury was 17% among all patients but significantly higher in non-survivors (59% versus 1%, p<0.0001). Another study reported the incidence of acute cardiac injury to be 7% (10 out of 138 patients), but significantly higher among patients requiring intensive care unit (ICU) admission (22.2% versus 2%, p<0.001).1 A meta-analysis of cardiac biomarkers in patients with COVID-19 showed that the values of cardiac troponin I were significantly higher in those with severe disease than in those without (SMD 25.6 ng/l; 95% CI [6.8–44.5 ng/l]).4

Hypercoagulability has also been reported among COVID-19 patients, which makes them more prone to arterial thrombi and ACS.12 In a recent review of COVID-19 patients, Violi et al. suggested that patients with severe disease (Pneumonia Severity Index >90, CURB-65 ≥2, acute respiratory distress syndrome, sepsis or requiring ICU admission) and those with elevated D-dimer levels have a hypercoagulable milieu and are likely to benefit from antithrombotic therapy with low molecular weight heparin or aspirin.13
It has been postulated that COVID-19 may cause direct vascular injury, as reported in a case report of right coronary artery subintimal haematoma associated with spontaneous coronary dissection.14 In a recently published case series of COVID-19 patients, Varga et al. found evidence of direct viral infection of the endothelial cells and resultant endotheliitis.15 Such endothelial inflammation would certainly predispose COVID-19 patients to ACS, especially given their hypercoagulable state. Although no studies have reported the precise incidence of ACS and acute MI with COVID-19, these conditions have been known to occur after severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) and influenza infections.16–18
Myocarditis
Myocarditis is another potential aetiology of acute myocardial injury in COVID-19 patients, although there remains a paucity of literature on cases confirmed with imaging or histopathology.
SARS-CoV-2 can directly infect the cardiac tissue via angiotensin-converting enzyme 2 (ACE-2) receptors, which may cause myocardial inflammation and damage. Such a mechanism of myocardial injury through SARS-CoV has been well-established in animal models and cardiac autopsies.19,20 Some autopsy reports of COVID-19 patients have likewise reported myocardial interstitial infiltration by mononuclear cells and lymphocytic infiltration.21,22 Endomyocardial biopsy showing low-grade myocardial inflammation and viral infiltration directly into the myocardial tissue has also been reported.23
Isolated cardiac involvement with no respiratory symptoms or infiltrate has also been reported with cardiac MRI demonstrating marked biventricular myocardial interstitial oedema with late gadolinium enhancement.24,25 COVID-19-associated fulminant myocarditis has also been reported.26 As most patients do not undergo advanced cardiac imaging or cardiac biopsies (because of concerns of field contamination and increased viral dissemination), the true incidence of myocarditis remains unknown.
In addition to myocardial inflammation, pericardial involvement in the form of pericardial effusion with tamponade has also been reported in COVID-19.27 A mimicker of COVID-19-associated myocarditis is stress-induced cardiomyopathy, which is another known cause of acute heart failure in these patients.28
The exact aetiology of myocarditis in COVID-19 remains unknown. Possible mechanisms include direct viral infiltration and resultant inflammation, or a cytokine storm with myocardial inflammation.
Role of Cytokine Storm
An excessive immune response to SARS-CoV-2 infection has been demonstrated in certain subgroups of infected patients and is referred to as a cytokine storm.29,30 This phenomenon has been demonstrated in previous studies where SARS-CoV-1 caused severe lung injury.31
A cytokine storm is triggered by an imbalanced response of type 1 and type 2 T helper cells, resulting in an excessive production of cytokines, particularly interleukin 6 (IL-6).32,33 Yang et al. showed that in 53 clinically moderately to severely affected COVID-19 patients, 14 cytokines were elevated, with three of them – interferon gamma induced protein 10 (IP-10), monocyte chemotactic protein 3 (MCP-3) and interleukin-1 receptor antagonist (IL-1ra) – being independently associated with hypoxaemia, disease progression and death.34
The cytokine profile in COVID-19 is similar to that in secondary haemophagocytic hymphohistiocytosis (SHH), a hyper-inflammatory syndrome seen in other viral infections.35,36 The inflammatory response may impact myocardial contractility and function by direct myocardial damage or via hypoxia-mediated myocardial damage.37 Certain anti-cytokine treatments, such as tocilizumab, have been proposed to treat this cytokine storm.38
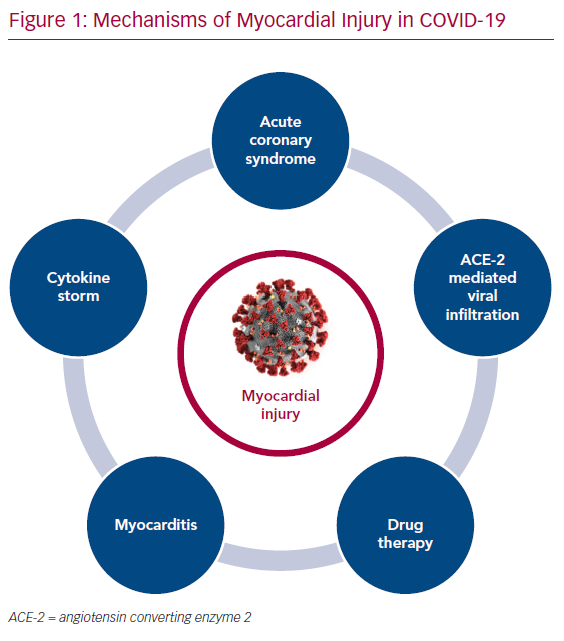
Role of ACE Receptor in Pathogenesis
ACE-2 is a membrane-bound aminopeptidate receptor expressed on the epithelial cells of the lungs, intestines, kidneys and blood vessels.39 It has important immune and cardiovascular roles. Angiotensin-converting enzyme (ACE) cleaves angiotensin I to generate angiotensin II (Ang II), which binds to and activates AT1R, thus promoting vasoconstriction. ACE-2 cleaves angiotensin II and generates angiotensin 1–7, a powerful vasodilator acting through Mas receptors.
SARS-CoV-2 has a spike protein receptor-binding domain, similar to SARS-CoV-1, which interacts with the ACE-2 receptor and acts as the primary functional receptor for pathogenicity and human-to-human transmission.40 Furthermore, SARS-CoV-2 binding to ACE-2 leads to its downregulation and increases angiotensin II. This subsequently leads to lower amount of angiotensin 1–7. This causes AT1R-mediated pulmonary vascular permeability.41
The ACE-2 receptor is upregulated in patients with cardiovascular disease, especially with the use of renin-angiotensin-aldosterone system inhibitors.32 SARS-CoV-2 mainly affects the alveolar epithelial cells, resulting in respiratory symptoms, which are more severe in patients with cardiovascular disease.
Some animal studies have reported the protective role of ACE-2 in the development of severe acute lung injury.42 This has led to speculation about the potential effects of antihypertensive medications with ACE-inhibitors or angiotensin receptor blockers on COVID-19 positive patients.
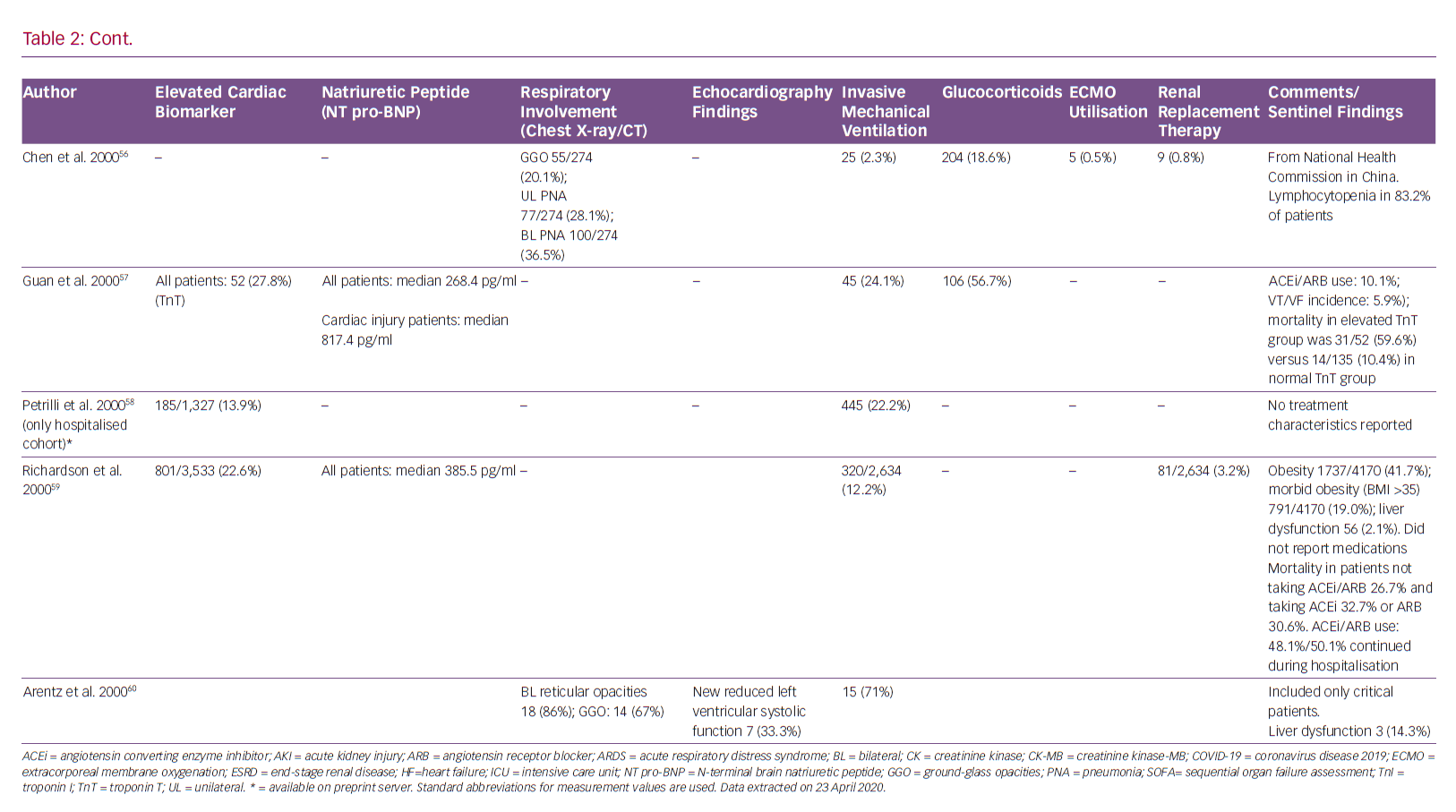
Guo et al. demonstrated that angiotensin-converting enzyme inhibitors/angiotensin receptor blockers had no effect of on mortality in 187 hospitalised patients with SARS-CoV-2.7 Furthermore, a retrospective analysis of 1,128 adult COVID-19 patients with a previous history of hypertension, inpatient use of ACE inhibitors or angiotensin receptor blockers (ARBs) was associated with lower risk of all-cause mortality compared to non-users.21 There is a lack of clear experimental or clinical data showing ACE receptor-mediated antihypertensive therapy has adverse effects in patients with COVID-19.43 Therefore, modification of pre-existing therapy is not recommended as per guidelines from both American and European cardiovascular societies.44,45
On the other hand, sacubitril/valsartan reduces the concentration of pro-inflammatory cytokines and neutrophil count, while increasing lymphocyte count more than valsartan alone or placebo. This finding might be related to the increase in plasma levels of atrial/brain/C-type natriuretic peptide, Ang I/II, substance P, bradykinin and endothelin secondary to neprilisin inhibition by sacubitril.46 Therefore, administration of an ACE, an ARB or sacubitril/valsartan may even be beneficial through inhibition of AT1R.47
Impact of Drug Therapy
Currently, there is no known no curative therapy for SARS-CoV-2. Many drugs are being used as prophylaxis or for treatment of COVID-19 patients in an expedited manner.48 Hydroxychloroquine, proposed as an effective strategy for COVID-19 patients, has known cardiovascular toxicity, causing arrhythmias and biventricular failure.5,49
Hydroxychloroquine-related cardiac adverse events are rare but can be severe and, occasionally, life-threatening. A recent review of cardiac complications attributed to this medication found that most patients who developed cardiac symptoms had been on treatment for a long period of time (median 7 years), and had been exposed to large cumulative doses (median 1,235 g.50 Conduction abnormalities (prolonged QT and PR intervals) were the most common adverse events, affecting 85% of these patients. Although it appears that conduction abnormalities are a long-term consequence of high-dose and prolonged use of hydroxychloroquine, we recommend monitoring all patients with COVID-19 treated with this drug for cardiac arrhythmias.
Azithromycin, which has been proposed for the treatment of COVID-19 pneumonia, is also known to increase the risk of adverse cardiovascular events. This risk is highest among patients with baseline cardiovascular comorbidities, conduction abnormalities and on concomitant QT-prolonging medications.51 Indeed, concomitant use of hydroxychloroquine and azithromycin is associated with higher risk of QTc prolongation in COVID-19 patients than either medication alone.52
The potential of experimental treatment to cause myocardial damage remains a concern and patients receiving such therapies require close monitoring.
Long-term Cardiovascular Implications
In addition to acute myocardial damage, COVID-19-related long-term cardiovascular morbidity is also a concern. SARS-CoV-2 is structurally and genetically very similar to its predecessor SARS-CoV-1.
A small study of 25 patients who recovered from SARS-CoV-1 infection had abnormal lipid profiles and glucose metabolism, and a higher burden of cardiovascular abnormalities at 12 years’ follow-up.53 As serological antibody testing becomes readily available to identify patients who have recovered from COVID-19, it will be important to observe their long-term cardiovascular health after infection.
Conclusion
Multiple mechanisms appear to contribute to myocardial injury in COVID-19. Individually or in conjunction with one another, they include respiratory failure induced hypoxia, inflammatory cytokine storms, direct viral infiltration and subsequent myocyte death, and myocardial dysfunction from acute illness.