








Chronic heart failure (HF) is a complex and progressive clinical syndrome resulting from any abnormality of cardiac structure or function. The American College of Cardiology Foundation/American Heart Association guideline defines HF as ‘a complex clinical syndrome that results from any structural or functional impairment of ventricular filling or ejection of blood’.1 The European Society of Cardiology definition is ‘an abnormality of cardiac structure or function leading to failure of the heart to deliver oxygen at a rate commensurate with the requirements of the metabolising tissues, despite normal filling pressures (or only at the expense of increased filling pressures).2
HF prevalence and the number of HF-related hospitalisations are increasing, and the prognosis remains poor, with a 5-year mortality worse than many cancers.3,4 There has been significant progress in HF therapy, but mostly in HF with reduced ejection fraction (HFrEF), while for patients with preserved ejection fraction (HFpEF), no therapy has improved clinical outcomes.2,5 Despite such advances, however, morbidity and mortality of HFrEF still remains high. It is evident, therefore, that substantial unmet needs exist in HF therapy. This article aims to review the mechanism of action and clinical development of sacubitril/valsartan (LCZ696), a first-in-class angiotensin receptor neprilysin inhibitor that has recently received regulatory approval in the US and Europe.
The Role of the Renin-Angiotensin-Aldosterone System in Heart Failure
Neurohumoral activation, in particular, of the renin-angiotensinaldosterone system (RAAS) and the sympathetic nervous system, plays a major role in the development and progression of HF.1,2 The RAAS is an essential component in the regulation of cardiovascular homeostasis that exerts its actions through the hormones angiotensin II and aldosterone. The RAAS regulates vascular tone and blood pressure (BP) by means of vasoconstriction and renal sodium and water retention.6 Abnormalities in cardiac function in HF activate the RAAS and sympathetic nervous system in order to maintain perfusion of vital organs.7 However, prolonged activation of these systems increases systemic vascular resistance and causes sodium and water retention, myocardial hypertrophy, fibrosis and apoptosis, which accelerates the progression of HF and promotes end-organ damage.6,8–10
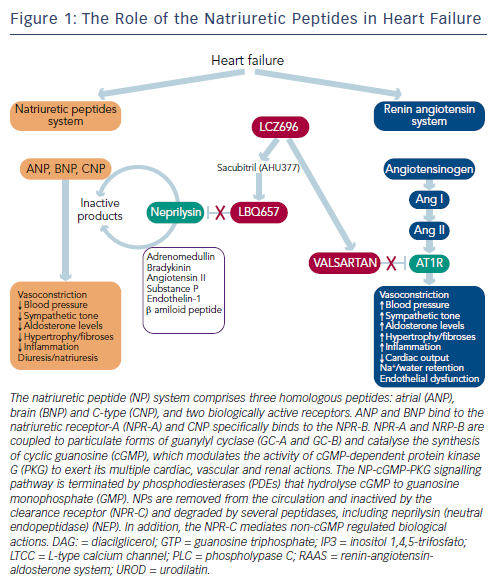
The blockade of beta-adrenergic receptors leads to symptomatic improvement and reduced morbidity and mortality in patients with HFrEF.9–13 In addition, the central role of the RAAS system in HF has led to the therapeutic use of RAAS inhibitors,2,8 including angiotensin-converting enzyme (ACE) inhibitors,14 angiotensin receptor blockers (ARBs) in patients who cannot tolerate ACE inhibitors15 and mineralocorticoid receptor antagonists in the treatment of chronic HF.16,17 ARBs competitively inhibit the binding of angiotensin II to its AT1 receptors located on blood vessels and other tissues, and improve symptoms, haemodynamics and outcomes in chronic HF.1,2 These beneficial effects are attributed to the inhibition of the deleterious effects of AT1 receptor stimulation, i.e., vasoconstriction, Na+ and water retention, aldosterone and vasopressin release, stimulation of sympathetic tone, inflammation, fibrosis and cell growth (see Figure 1).
However, ACE inhibitors, ARBs, aldosterone receptor antagonists and combinations of drugs in these classes are limited in their ability to fully inhibit the activity of the RAAS.6,18 Furthermore, ACE inhibitors and ARBs induce a reactive rise in plasma renin activity that may eventually surpass their RAAS-inhibitory effect, and plasma aldosterone levels remain elevated in a subset of patients despite therapy, a phenomenon known as aldosterone escape or aldosterone breakthrough.19 In addition, ARBs do not enhance bradykinin-mediated vasodilation and are considered less effective than ACE inhibitors.2
For the past 25 years, an add-on therapy approach to chronic HF has been used, beginning with diuretics, then adding ACE inhibitors (or ARBs) and beta blockers, followed by mineralocorticoid receptor antagonists.13,20,21 Ivabradine, which reduces heart rate, is also approved as an add-on therapy in HF.22 Nevertheless, morbidity and mortality remain high and there is, therefore, a need for new therapeutic targets in HF.
Role of Natriuretic Peptides in Heart Failure
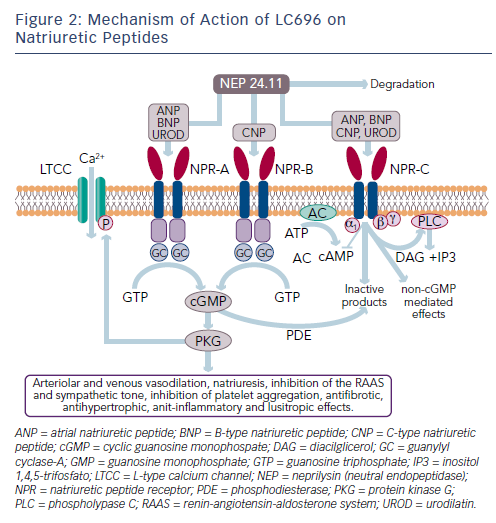
While the activation of the RAAS and sympathetic nervous system is detrimental in HF, other counter-regulatory pathways are activated in HF, including the natriuretic peptide (NP) system (see Figure 2). The NP system consists of atrial (ANP),23 B-type (BNP)24 and C-type (CNP) NPs; these hormones regulate BP and fluid homeostasis.25–27 ANP is synthesised and secreted in atria, BNP is secreted from the ventricles in response to mechanical stretch and increased intracardiac volume/ pressure and CNP mostly originates from endothelial and renal cells and is secreted in response to endothelium-dependent agonists and pro-inflammatory cytokines.25,26,28 NPs activate three transmembrane receptors: natriuretic peptide receptor (NPR)-A, NPR-B and NPR-C.27 The binding of NPs to type A (NPR-A) and type B (NPR-B) receptors activates guanylate cyclase, increasing levels of the second messenger cyclic guanosine monophosphate (cGMP) and its effector molecule protein kinase G. This induces natriuresis, diuresis, vasodilation and inhibition of the RAAS system and the sympathetic nervous system, as well as antifibrotic, antiproliferative and antithrombotic effects (see Figure 2).25,26,28
A growing body of evidence suggests that hypertension and HF may be consequences of a dysregulated NP system and that patients with HF and hypertension may have a deficiency of biologically active NPs.28,29 NPR-C clears NP from the circulation through receptormediated internalisation and degradation. Urodilatin, a renally synthesised isoform of ANP, stimulates NPR-A located in the glomeruli and collecting ducts and promote Na+ excretion.30
Another key component of the NP system is neprilysin (neutral endopeptidase 24.11), which catalyses the degradation of ANP, BNP and CNP as well as the degradation of bradykinin, adrenomedullin, endothelin-1, substance P and angiotensin II (see Figure 1).31 Neprilysin is a potentially useful therapeutic target in HF.6,28 Inhibition of neprilysin increases the levels of NP, causing vasodilation and a reduction in extracellular fluid volume. Neprilysin does not hydrolyse N-terminal prohormone of brain NP (NT-proBNP), therefore the latter is a useful cardiac biomarker to assess therapeutic effect and prognosis in patients treated with neprilysin inhibitors.32
Clinical Development of Vasopeptidase Inhibitors
Augmentation of NPs by direct administration of these peptides is difficult because oral delivery is ineffective and parenteral delivery problematic. While nesiritide has been shown to produce a modest improvement in dyspnoea, it does not favourably affect clinical outcomes, decongestion or renal function33–35 and safety concerns have been raised.36,37 Blockade of NP breakdown by neprilysin inhibitors has, therefore, been investigated.38 Oral neprilysin inhibitors, such as candoxatril, produced clinical benefit in patients with chronic HF.39,40 However, candoxatril has no effect on, or increases, systolic BP (SBP) in normotensives, an effect prevented by enalapril, and does not reduce BP in hypertensive subjects, probably because its vasodilatory effect may be offset by an increased activity of the RAAS and sympathetic nervous system and/or by downregulation of NP receptors.41,42 In addition, since neprilysin acts on numerous physiological targets, the effect of candoxatril was broader than anticipated.41
Neprilysin inhibition results in activation of the RAAS, therefore, in order to be clinically beneficial, neprilysin inhibition requires concomitant inhibition of the RAAS.43 Vasopeptidase inhibitors are dual inhibitors of ACE and neprilysin and, therefore, emerged as a new therapeutic option in HF and hypertension, but their pharmacological profile is complex.44 Omapatrilat was more effective than either lisinopril or amlodipine in reducing BP,44 but in patients with chronic HF it was not more effective than enalapril in reducing the combined risk of death or hospitalisation for HF requiring intravenous treatment.45 However, omapatrilat was discontinued due to the risk of angioedema, possibly due to excessive inhibition of bradykinin degradation (presumably via neprilysin, ACE and aminopeptidase P).46,47
Mechanism of Action of LCZ696
Following the disappointing outcomes of combined ACE/neprilysin inhibition, the combination of neprilysin and an ARB was investigated. ARBs have a lesser effect on bradykinin48 and have been associated with lower risk of angioedema compared with ACE inhibitors, not significantly different from placebo.49,50 Therefore, RAAS blockade at the AT1 receptor appears to be a preferable strategy to ACE inhibition.51 LCZ696 (Entresto™, Novartis) is a first-in-class angiotensin II receptorneprilysin inhibitor (ARNI) whose multimodal mode of action involves neprilysin inhibition and AT1 receptor blockade. LCZ696 is composed of two molecular moieties (in a 1:1 molar ratio) in a single crystalline complex comprising valsartan (an ARB) and sacubitril (AHU377).52 After ingestion, LCZ696 undergoes rapid dissociation into valsartan and sacubitril, a prodrug that is subsequently de-ethylated by esterases to LBQ657, a neprilysisn inhibitor.
In healthy volunteers, LCZ696 causes dose-dependent increases in ANP, plasma and urinary cGMP, renin concentration and activity and angiotensin II levels, as a result of neprilysin inhibition and AT1 receptor blockade (see Figure 1).52 After LCZ696 administration, levels of cGMP significantly increased at 4 and 12 hours and returned to baseline levels at 24 hours; all RAAS biomarkers reached a maximum after 4 hours and remained elevated at 24 hours.52 Thus, the pharmacodynamic effects of valsartan and LBQ657 are similar. In HF patients, levels of urinary cGMP, plasma BNP and renin concentration and activity were higher during treatment with LCZ696 than with enalapril, while circulating levels of markers of myocardial wall stress (N-terminal pro-BNP) and myocardial injury (troponin T) were lower during treatment with LCZ696 than with enalapril.53,54 After 21 days of LCZ696 administration (100 mg titrated to 200 mg daily) a significant lowering of plasma NT-proBNP, aldosterone and endothelin-1 levels was observed.55
Nevertheless, the precise mechanism by which LCZ696 reduces cardiovascular mortality in HF patients is uncertain.56 The observed benefit is likely to be related to the incremental benefits of neprilysin inhibition, which may counteract the detrimental effects of RAAS and sympathetic nervous system activation. Other possible mechanisms might include: a haemodynamic improvement related to NP-mediated reduction in ventricular wall stress; an improvement in ventricular function; modification of the basis for fatal ventricular arrhythmias via decreased myocardial fibrosis and hypertrophy, attenuation of ventricular remodelling or direct anti-arrhythmic properties; sympatholytic or vagotonic effects of hormones potentiated by neprilysin inhibition; and anti-atherosclerotic or anti-thrombotic effects of enhanced NP expression leading to an improvement in regional myocardial perfusion.56 Further understanding of the mechanisms of action of LCZ696 would provide a deeper insight into the pathophysiology of HF and should be a priority in the future.
Pharmacokinetic Properties of LCZ696
Valsartan and LBQ657 have similar pharmacokinetic profiles, with rapid absorption, reaching maximum plasma concentrations within 1.5–4.5 hours, and a half-life of 8.9–16.6 and 9.9–11.1 hours, respectively, indicating that both agents exhibit comparable pharmacokinetic properties and that LCZ696 was suitable for once- or twice-daily dosing.52 Sacubitril (AHU377) reaches peak plasma levels within 0.5–1.1 hours and presents a half-life of 1.1–3.6 hours owing to its rapid conversion into the active metabolite LBQ657, which explains the rapid onset of activity of LCZ696. LCZ696 achieves 90 % inhibition of neprilysin.52 Importantly, LCZ696 does not inhibit aminopeptidase A, unlike omapatrilat, thus minimising the risk of angiodema.52 In healthy volunteers, LCZ696 400 mg and valsartan 320 mg provide similar exposure to valsartan.
Clinical Development of LCZ696
Heart Failure
The main clinical trials investigating the efficacy and safety of LCZ696 are summarised in Table 1. The pivotal clinical trial in the development of LCZ696 was the Prospective comparison of ARNI with ACEI to Determine Impact on. Global Mortality and morbidity in Heart Failure (PARADIGM-HF) study, which recruited patients (n=8,442) with chronic HF (New York Heart Association [NYHA] class II–IV) and reduced left ventricular ejection fraction (LVEF; ≤40 % to ≤35 %).57 The trial began with a single-blind run-in period to test drug tolerability. Patients received enalapril 10 mg twice daily for 2 weeks, then 100 mg LCZ696 twice daily for 1–2 weeks and then 200 mg twice daily for 2–4 weeks. Two brief (one day) washout periods were also included to minimise the potential risk of angioedema due to overlapping ACE and neprilysin inhibition. During the run-in period, 12 % of patients withdrew due to an adverse event. Following the run-in period, patients underwent double-blind 1:1 randomisation to LCZ696 200 mg twice daily or enalapril 10 mg twice daily. Exclusion criteria included symptomatic hypotension (SBP <100 mmHg) (at screening or 95 mmHg at randomisation), an estimated glomerular filtration rate (eGFR) <30 ml/min/1.73 m2 at screening or at randomisation or a decrease in the eGFR >25 % (which was amended to 35 %) between screening and randomisation, a serum K+ level >5.2 mmol/L at screening (or >5.4 mmol/L at randomisation) or a history of angioedema or unacceptable side effects during ACE inhibitor or ARB therapy.
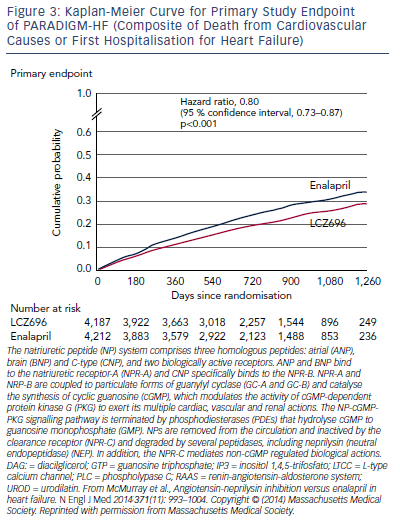
The trial was terminated early after a median follow up of 27 months due to evidence of an overwhelming benefit with LCZ696. The primary endpoint, a composite of death from cardiovascular causes or hospitalisation for HF, occurred in 21.8 % of the LCZ696 group and 26.5 % of the enalapril group (hazard ratio [HR] in the LCZ696 group, 0.80; 95 % confidence interval [CI], 0.73–0.87; p<0.001) (see Figure 3). During the trial, the numbers of patients who would need to have been treated to prevent one primary event and one cardiovascular death were 21 and 32, respectively. Death from any cause was reported in 17 % of patients receiving LCZ696 and 19.8 % receiving enalapril (HR 0.84; 95 % CI 0.76–0.93; p<0.001); of these 13.3 % and 16.5 %, respectively, died from cardiovascular causes (HR 0.80; 95 % CI 0.71–0.89; p<0.001).57 Using actuarial estimates from the PARADIGM-HF trial, and assuming that the protective effects of LCZ696 are sustained during long-term use, it has been estimated that treatment with LCZ696 could result in 1 to 2 years of increased life expectancy in patients with HF.58 LCZ696 also reduced the risk of hospitalisation for HF by 21 % compared with enalapril (p<0.001) and decreased HF symptoms (p=0.001).57 Interestingly, these benefits were also observed in 2,907 patients with diabetes. In a sub-analysis of PARADIGM-HF, the benefit of LCZ696 compared with enalapril was consistent, regardless of glycaemia status.59
In addition, two recent analyses have focused on the effect of LCZ696 on the risk of clinical deterioration. A subanalysis of PARADIGM-HF focused on pre-specified measures of non-fatal clinical deterioration.53 Compared with enalapril, fewer patients in the LCZ696 group required treatment intensification for HF (520 versus 604; HR 0.84; 95 % CI 0.74–0.94; p=0.003) or an emergency department visit for worsening HF (HR 0.66; 95 % CI 0.52–0.85; p=0.001). Patients receiving LCZ696 had 23 % fewer hospitalisations for worsening HF (851 versus 1,079;95 % CI 0.67–0.85; p<0.001) and 18 % fewer stays in intensive care (768 versus 879; p=0.005), and were 31 % less likely to receive intravenous positive inotropic agents (p<0.001) and 22 % less likely to have cardiac transplantation or implantation of a cardiac device for HF (p=0.07). The reduction in hospitalisation was noted within the first 30 days after randomisation. Worsening symptoms were consistently more commonly reported in the enalapril group.53
Another analysis focused on the mode of death in the PARADIGM-HF trial. The majority of deaths were cardiovascular (80.9 %), and treatment with LCZ696 significantly reduced the risk of cardiovascular death (HR 0.80; 95 % CI 0.72–0.89; p<0.001). This reduced risk was primarily due to a reduction in both sudden cardiac death (HR 0.80; 95 % CI 0.68–0.94; p=0.008) and death due to worsening HF (HR 0.79; 95 % CI 0.64–0.98; p=0.034). The treatment effect on sudden cardiac death was not affected by the presence or absence of an implantable cardioverterdefibrillator. 56 Of note, LCZ696 reduced cardiovascular death to a similar extent as its reduction of HF hospitalisation, while the results of many pivotal studies of RAAS in HF found a more pronounced reduction in hospitalisations for worsening HF than cardiovascular death.56
The Safety and Tolerability of Initiating LCZ696 in Heart Failure Patients (TITRATION) study demonstrated the safety and efficacy of up-titrating LCZ696 from 50 mg twice daily to a target dose of 200 mg twice daily in a 3- (condensed) versus 6-week (conservative) regimen in patients with HFrEF (EF ≤35 %) on beta-blockers. The study enrolled a broader range of patients than PARADIGM-HF, including inpatients and patients naïve to ACE inhibitors or ARBs.60 The study involved an open-label run-in period in which LCZ696 was tested for tolerability and safety at a 50 mg twice daily for 5 days. Patients were then randomised to up-titration of LCZ696 to 200 mg during the next 3 (condensed) or 6 weeks (conservative) regimen. Primary endpoints included the proportion of patients experiencing pre-specified adverse events (symptomatic hypotension, hyperkalaemia, renal dysfunction, angioedema) and outcomes including SBP <95 mmHg and a doubling of serum creatinine from baseline. In the primary endpoint of tolerability, there were no differences between groups. Treatment was successful in 78 % and 84 % of patients in the condensed and conservative regimens, respectively (p=0.07). The target dose was achieved and maintained for 12 weeks in 76 % of patients. The study also suggested that patients on ACE inhibitors or ARBs should probably be moved less quickly to up-titration of LCZ696.
There is a lack of effective treatments for patients with HFpEF, therefore LCZ696 was evaluated in this treatment setting. Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fraction (PARAMOUNT) was a phase II study in patients with NYHA class II–III HF and LVEF ≥45 %. Participants (n=301) were randomised to LCZ696 (titrated to 200 mg twice daily) or valsartan (titrated to 160 mg twice daily). The primary endpoint was change in NT-proBNP, a marker of left ventricular wall stress.61 At 12 weeks, NT-proBNP was significantly reduced in the LCZ696 group compared with the valsartan group (from 783 pg/ml to 605 pg/ml in the LCZ696 group versus from 862 pg/ml to 835 pg/ml in the valsartan group; ratio LCZ696:valsartan, 0.77; 95 % CI 0.64–0.92; p=0.005). In addition, after 36 weeks more patients in the LCZ696 group showed improvements in NYHA functional class and reduced left atrial size compared with valsartan, consistent with reverse left atrial remodelling. LCZ696 was well tolerated with adverse effects similar to those of valsartan. A recently reported analysis from this study investigated the effects of LCZ696 on renal function in patients with HFpEF. Treatment with LCZ696 for 36 weeks resulted in lower serum creatinine, higher eGFR and an increase in urinary albumin to creatinine ratio compared with valsartan.62
Although the PARAMOUNT study was not powered to detect clinical outcomes, it was a hypothesis-generating trial that provided the basis for the ongoing phase III Prospective comparison of Angiotensin Receptor neprilysin inhibitors with Angiotensin converting enzyme inhibitors to Determine Impact on Global Mortality and morbidity in Heart Failure (PARAGON-HF) trial, which aims to enrol 4,300 patients. Enrolment criteria are symptomatic HFpEF, NYHA class II–IV, LVEF ≥45 % requiring treatment with diuretics for HF ≥30 days prior to study entry, structural heart disease (left atrial enlargement or left ventricular hypertrophy) documented by echocardiogram, a HF hospitalisation within 9 months prior to study entry and/or an elevated NT-proBNP.63 The primary endpoint is a composite of cardiovascular death and total HF hospitalisations. The treatment arm with the lower rate of events will be deemed to have the most successful response.
Arterial Hypertension
LCZ696 has also shown efficacy in clinical studies of hypertension. In a multinational study with sites in 18 countries, patients (n=1,328) with mild-to-moderate hypertension were randomised to 8 weeks’ treatment with LCZ696 (100, 200 or 400 mg daily), valsartan (80, 160 or 320 mg daily), sacubitril (200 mg daily) or placebo.64 The reduction in mean resting SBP and diastolic BP (DBP) was significantly greater for 200 mg LCZ696 versus 160 mg valsartan (-11/-6.1 versus -5.7/-3.2 mmHg; p<0.001) and for 400 mg LCZ696 versus 320 mg valsartan (-12.5/-6.9 versus -6.4/4.1 mmHg; p<0.005).64 Response rates were also significantly higher in patients on 200 mg LCZ696 versus 160 mg valsartan (91/163, 56 %; p=0.0095), and on 400 mg LCZ696 versus 320 mg valsartan (103/163, 63 %; p=0.026). No differences were found between 100 mg LCZ696 and 80 mg valsartan. In a multicentre study carried out in Japan, China, South Korea, Taiwan and Thailand, patients aged ≥18 years (n=389) with hypertension were randomised to LCZ696 (100, 200 or 400 mg once daily once daily) or placebo (n=92) for 8 weeks. Reductions in SBP, DBP (p<0.0001) and pulse pressure (p<0.001) were significantly greater with all doses of LCZ696 than with placebo. The reductions are greater than those observed in white European populations, as this Asian population has a higher salt intake and increased salt sensitivity. There were also significant reductions in 24-hour, daytime and nighttime ambulatory SBP, DBP and pulse pressure for all doses of LCZ696 compared with placebo (p<0.0001).65 Although the mechanism is uncertain, this effect can be related to its vascular effects and/or to reduced effective circulating volume. In the PARAMOUNT trial the reduction from baseline in mean SBP/DBP was -9.3/-4.9 mmHg with LCZ699 (200 mg twice daily) and -2.9/-2.1 mmHg with valsartan (160 mg twice daily).61 In the PARADIGMHF trial where 71 % of patients had hypertension, LCZ696 therapy resulted in a significant reduction in SBP compared with enalapril (mean difference -2.7 mmHg; p<0.001) over a period of 3 years. The role of the reduction in SBP on the decreased rate of death and HF hospitalisation observed in this trial is uncertain.
Safety Profile of LCZ696
In clinical studies of hypertension, LCZ696 was well-tolerated and no cases of angioedema or deaths were reported. The most common adverse events were headache and pruritus,64 nasopharyngitis and upper respiratory tract infection.64,65 Hypotension or syncope occurred in five patients (one each in the placebo, 400 mg LCZ696 and 200 mg AHU377 groups; two in the 200 mg LCZ696 group).64 Adverse events resulting in treatment discontinuation occurred in 1–2 % of patients on LCZ696, with the highest occurrence in the AHU377 and placebo groups.64 However, patients with diabetes and renal disease (eGFR >30 ml/min/1.73 m2) were excluded from these trials.
In the PARADIGM-HF study, 12 % of patients did not complete the run-in period because of adverse events, most frequently cough, hyperkalaemia, renal dysfunction or hypotension. Across the study period, LCZ696 was discontinued in 746 patients (17.8 %) and enalapril in 833 patients (19.8 %). During the double-blind phase, the LCZ696 group had higher rates of hypotension (p<0.001) and non-serious angioedema (p=0.31), but significantly lower rates of serum creatinine ≥2.5 mg/dl, serum potassium ≥6 mmol/L and cough than the enalapril group, and a lower overall incidence of adverse events. Fewer patients in the LCZ696 group than in the enalapril group stopped drug medication because of an adverse event (10.7 % versus 12.3 %; p=0.03) or renal impairment (0.7 % versus 1.4 %; p=0.002).57 In the PARAMOUNT trial the number of patients with hypotension, renal dysfunction or hyperkalemia did not differ between groups.61
However, questions remain regarding the safety of LCZ696. Although it is better tolerated than valsartan in clinical studies, it showed a higher incidence of hypotension, an important consideration in elderly patients, although this rarely resulted in discontinuation of treatment, and the number of discontinuations due to hypotension were balanced across both groups. In addition, the question of angioedema in daily clinical practice remains unanswered. Angioedema is more common in patients of African origin,66,67 but these were under-represented in the PARADIGM-HF (5 %). There is also a need for studies in ACE inhibitornaïve patients, where benefits were less pronounced in PARADIGM-HF, although it should be noted that all patients had an enalapril run-in phase so were not truly naïve.
A recent review paper discussing preclinical models and human genetic analyses suggested that neprilysin inhibitors may lead to an accumulation of amyloid beta-peptide in the brain and may thus accelerate Alzheimer’s disease progression in at-risk patients.68 This is a hypothetical concern and not based on any human studies: a Chinese study found no association between two NEP gene polymorphisms and Alzheimer’s disease in elderly people.69 Furthermore, a 2-week LCZ696 administration in human healthy volunteers did not modify Abeta1−40 and Abeta1−42 levels in the cerebrospinal fluid70 and no cognition-related adverse events related to treatment have been reported in any of the randomised clinical trials to date, probably because multiple (20) proteins are involved in the clearance of amyloid beta-peptides. There is therefore no conclusive evidence for an association between NEP and Alzheimer’s disease in humans. Cognition-related adverse effects were observed in the PARADIGM-HF trial, as expected in a study population including elderly patients, but the incidence was balanced in both treatment arms.71 Serial cognition testing will be performed in PARAGON-HF.63 In addition, a dedicated study investigating cognition and PET imaging is planned.
Although clinical trials to date have demonstrated the tolerability of LCZ696, it can be argued that the similar rates of adverse events among LCZ696 and standard therapy may have been the result of patient selection bias. Thus, data from on-going clinical trails and clinical practice are needed to evaluate its long-term efficacy.
Clinical Use of LCZ696
LCZ696 has been approved by the European Medicines Agency72 and US Food and Drug Administration73 to reduce the risk of cardiovascular death and hospitalisation in adult patients with symptomatic chronic HFrEF. It is usually administered in conjunction with other HF therapies, in place of an ACE inhibitor or an ARB. If switching from an ACE inhibitor, a washout period of 36 hours is important. The recommended starting dose is 49/51 mg (sacubitril/valsartan) twice daily. This may be increased after 2–4 weeks to the target maintenance dose of 97/103 mg (sacubitril/ valsartan) twice daily as tolerated by the patient. The starting dose should be reduced to 24/26 mg (sacubitril/valsartan) twice daily for patients not currently taking an ACE inhibitor or an ARB, or previously taking a low dose of these agents, i.e. patients with severe renal impairment (eGFR <30 ml/min/1.73 m2) and patients with moderate hepatic impairment (Child-Pugh B).
Conclusion
While drugs targeting the RAAS represent the cornerstone of HF treatment, there is a need for novel therapeutic approaches. LCZ696 is an effective and safe alternative to ACE inhibitors and may change future first-line approaches to HF therapy because of its significant improvement in survival and reduced rates of rehospitalisation. Additionally, LCZ696 has been found more effective than valsartan in hypertensive patients, although comparative studies with other antihypertensive drugs are needed and its effects on cardiovascular outcomes are unknown. Ongoing clinical trials will define its future role in the treatment of HF and other cardiovascular diseases, where ACE inhibitors or ARBs are currently first-line therapies.
For LCZ696 to displace ACE inhibitors and ARBs in daily clinical practice, more information on real-life use of LCZ696 is required, including safety and efficacy in patient groups not included in PARADIGM-HF, i.e. patients with acute decompensated HF and more advanced symptoms (NYHA IV only represented 0.8 % of the study population); elderly and black patients; patients with resistant hypertension, nephropathy and proteinuric renal disease; patients receiving high doses (≥10 mg twice daily); or treated with ARBs. There is some preliminary evidence of LCZ696 in hypertensive patients with diabetes; furthermore, the benefit of LCZ696 in the PARADIGM-HF trial was reported in 2,907 patients with diabetes. It will also be important to determine which patients will benefit most, for example ACE inhibitor/ARB naïve patients, who are commonly encountered in the clinic. For the first time in 30 years, physicians must make a careful therapeutic decision: instead of adding to a drug regimen, they have the option to replace ACE inhibitors or ARBs with LCZ696. This decision should be made with the knowledge that the agent provides proven benefits in terms of reduced mortality and fewer re-hospitalisations.