










Patients with a broad range of systemic inflammatory rheumatic diseases (i.e. rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis) are exceptionally prone to cardiovascular disorders, but previous work concerning the development of heart disease in these individuals has largely focused on the risk of MI.
However, the most important cardiovascular complication in these individuals is the development of heart failure (HF), an event that may not be readily apparent to many clinicians and is not related to traditional cardiovascular risk factors or to clinically evident ischaemic heart disease.1,2 HF develops with increasing rapidity following a diagnosis of systemic rheumatic diseases, and the magnitude of risk is related to the severity of arthritic activity.2 Typically, the four principal systemic rheumatic disorders, that is, rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis, increase the risk of new-onset HF as much as two- to threefold, when compared with the general population.2–9
Importantly, both the clinical presentation and the outcome of HF differ between patients with and without these systemic rheumatic diseases. Although patients with systemic rheumatic diseases are prone to MI (a form of injury that is typically linked to HF with reduced ejection fraction [HFrEF]), the phenotype of HF in most patients with systemic rheumatic diseases is characterised by the absence of coronary artery disease (CAD) and a preserved EF, with its characteristic features of diastolic filling abnormalities, microvascular dysfunction and cardiac fibrosis on cardiac imaging.10–17 This finding is not surprising; of the two main phenotypes of HF, that is, HFrEF or HF with preserved EF (HFpEF), the former is related to cardiomyocyte loss and stretch, whereas the latter is primarily related to the presence of systemic inflammatory disorders, most commonly, obesity and diabetes.18–20
Mechanisms by Which Systemic Inflammatory Disorders Cause the Phenotype of HFpEF
Systemic inflammatory disorders can target the microcirculation of the myocardium, leading to microvascular endothelial dysfunction and fibrosis. The inflammatory injury can directly affect the coronary microvasculature; additionally, its effect can be mediated indirectly, because systemic inflammation promotes the expansion and biological transformation of epicardial adipose tissue (EAT), which acts as an amplifier to focus the systemic inflammatory process onto the underlying myocardium, with which it shares an unobstructed microcirculation.21,22 In healthy people, epicardial fat has the biological properties of brown adipose tissue, which combusts pro-inflammatory fatty acids and secretes adipokines (e.g. adiponectin) that suppress inflammation and nourish the heart. However, in the presence of systemic inflammation, mesenchymal cells in the epicardium proliferate and develop features of white adipose tissue, which is prone to lipolysis and causes the release of fatty acids that trigger infiltration of the tissues by macrophages, as well as the secretion of pro-inflammatory cytokines (e.g. leptin, tumour necrosis factor [TNF]-alpha, interleukin [IL]-6, IL-1beta and resistin).22–25
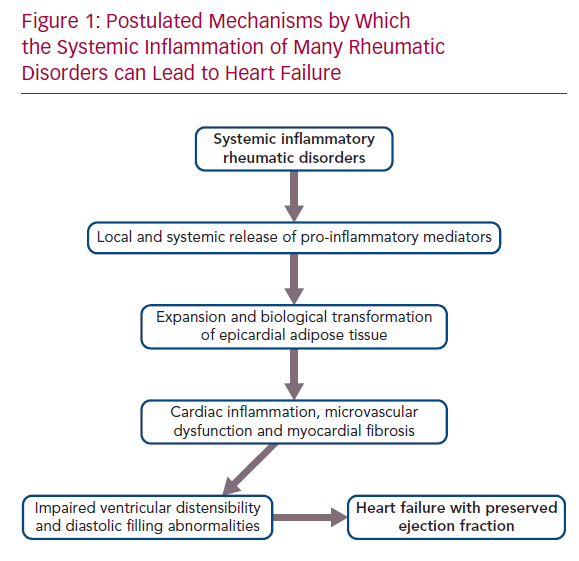
It is therefore noteworthy that EAT mass is increased in patients with rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis.26–30 The intimacy of its interface with the myocardium allows the biological derangements in epicardial fat to be readily transmitted to the neighbouring muscle.23 Acting locally, pro-inflammatory adipocytokines synthesised in and released from epicardial fat depots can cause a spread of the inflammatory process to underlying tissues, which includes the coronary arteries, as well as the atrial and ventricular myocardium, leading to the epicardial adipose inflammatory triad: coronary atherosclerosis, AF and HFpEF.31 The extension of inflammation to the perivascular tissues surrounding the coronary arteries likely explains the increased risk of MI in rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis.32–34 The spread of inflammation to myocardial tissues leads to microcirculatory dysfunction and rarefaction, as well as to increased deposition of collagen.35,36 MRI in patients with heightened quantities of epicardial fat reveals increases in extracellular volume, indicative of underlying myocardial fibrosis.36 If the process abuts the atria, the resulting electroanatomical remodelling and fragmentation leads to AF, thus explaining the increased incidence and prevalence of the arrhythmia in rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis.37–40 Finally, if the abnormalities in EAT adjoin the left ventricle, the microvascular dysfunction and cardiac fibrosis act together to impair the distensibility of the chamber, thereby limiting its ability to accommodate blood volume, but without a decrease in systolic function as measured by EF.41,42 The result is the HF phenotype known as HFpEF (Figure 1).
Mediating Effects of Leptin, Aldosterone and Natriuretic Peptides in the Pro-inflammatory Expansion of EAT
Why does systemic inflammation cause an expansion and pro-inflammatory transformation of EAT? The interactions among several endogenous hormonal mediators that are known to play a role in systemic inflammation (i.e. aldosterone, natriuretic peptides and leptin) contribute importantly to the development of epicardial adiposity and inflammation.4,5 Aldosterone promotes adipogenesis and causes an expansion and pro-inflammatory transformation of EAT,43,44 whereas epicardial adiposity is inversely related to the level of endogenous natriuretic peptides,45 presumably because natriuretic peptide signalling antagonises adipocyte hypertrophy.46 Additionally, aldosterone promotes pro-inflammatory pathways;47 mineralocorticoid receptor antagonism attenuates inflammasome activity and blocks the production of pro-inflammatory cytokines in adipocytes and macrophages.48 Conversely, endogenous natriuretic peptides also play an important role in the pathogenesis of systemic inflammatory disorders, but in a manner that opposes the actions of aldosterone. Natriuretic peptides inhibit biological pathways involved in inflammation and attenuate the production of pro-inflammatory cytokines in macrophages and adipocytes.49,50 Finally, EAT secretes leptin, which is also known to play a central role in immune responses and inflammation.51 Leptin mediates the proliferation of monocytes and their production of pro-inflammatory cytokines;51,52 circulating levels of the adipokine are elevated in systemic inflammatory states and are directly related to epicardial fat volume.53,54
Patients with obesity have increased circulating levels of aldosterone and leptin and decreased levels of natriuretic peptides.55–57 These relationships can be explained by the secretion of both aldosterone and leptin by an expanded mass of adipocytes,58 as well as by an ability of adipocytes to enhance the degradation of natriuretic peptides, either through an action to secrete neprilysin or to enhance peptide clearance.59,60 The increase in aldosterone and leptin (accompanied by the decrease in natriuretic peptides) acts to promote the systemic pro-inflammatory state. The interplay of these mediators may explain why obesity increases the incidence, enhances the clinical and radiographic severity and attenuates the degree of symptomatic improvement following the use of anti-inflammatory treatment in patients with rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis.61–68
In patients with expanded and dysfunctional EAT, the release of leptin and aldosterone can promote inflammation, microcirculatory dysfunction and fibrosis in the underlying myocardium,69 effects that are opposed by the anti-inflammatory and antifibrotic actions of endogenous natriuretic peptides.70 The result of these interactions is an impairment of the distensibility of the left ventricle, such that blood volume is accommodated only at the expense of a disproportionate increase in cardiac filling pressures, thus leading to exertional dyspnoea and HF. These observations explain why patients with HFpEF have elevated circulating levels of leptin and aldosterone but inappropriately suppressed levels of natriuretic peptides.25,42,71 Interference with the actions of aldosterone (and possibly leptin), as well as potentiation of natriuretic peptides, has yielded structural and clinical benefits in patients with HFpEF in randomised controlled trials.72–77
Activation of Pathogenetic Mechanisms for HFpEF in Systemic Rheumatic Diseases
Both rheumatoid arthritis and ankylosing spondylitis are characterised by increased levels of aldosterone in circulating blood and inflamed tissues,78,79 and spironolactone’s anti-inflammatory actions have been proposed as a treatment for these arthritides, as well as for lupus nephritis.80,81 At the same time, levels of leptin in blood (and often synovial fluid) are increased in rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis; are associated with the degree of joint and skeletal involvement; and are linked with patient-reported outcomes and measures of disease activity.82–93 Interestingly, high levels of leptin may identify patients with rheumatic diseases who respond poorly to anti-inflammatory treatment.94 Interestingly, increased leptin signalling may play a direct pathogenetic role in both rheumatoid arthritis and systemic lupus erythematosus by increasing the production of pro-inflammatory cytokines and autoantibodies and inhibiting immune regulation;95–99 interference with leptin signalling ameliorates the course of experimental lupus.100
Finally, the activity of neprilysin is increased at sites of joint involvement in rheumatoid arthritis,101 where it may play a role in degrading locally active natriuretic peptides and limiting their counterbalancing anti-inflammatory and anti-adipogenic actions. The release of neprilysin from the expanded mass of epicardial adipocytes may further diminish the ability of natriuretic peptides to minimise local cardiac fibrosis.102 This accelerated degradation of biologically active natriuretic peptides should not be confused with increased circulating levels of N-terminal pro-B-type natriuretic peptide (an inactive prohormone) in systemic rheumatic diseases; the prohormone is indicative of cardiac stress and is not degraded by neprilysin. As a result of these proadipogenic and pro-inflammatory interactions, EAT is increased in patients with rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis, and the magnitude of increase is generally proportional to the duration and severity of the underlying systemic inflammatory process.26–30
The transmission of epicardial inflammation to the underlying myocardial tissues explains why many patients with systemic rheumatic disease have coronary microcirculatory dysfunction, cardiac fibrosis, ventricular diastolic filling abnormalities, left atrial enlargement and HF in the absence of any evidence for or a history of MI or myocardial injury.1,11–17,26,103–110 Subclinical evidence for myocardial fibrosis on cardiac MRI and echocardiographic features of elevated cardiac filling pressures (as reflected by abnormal diastolic filling dynamics or abnormal left atrial geometry, and derangements in coronary microcirculatory dysfunction) are present in 30–50% of patients with rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis, particularly those with meaningful systemic inflammatory activity, and these cardiac structural and functional abnormalities parallel the severity and duration of joint involvement.26,105–107,110–112 These are the hallmarks of the ventricular myopathy that underlies the development of HFpEF, and they are often undiagnosed in clinical practice.
Effect of Anti-cytokine Agents and Neurohormonal Antagonists on the Development of HF In Systemic Rheumatic Disorders
If the proposed framework is valid, then interventions that directly interrupt inflammatory pathways might be useful in preventing and treating HFpEF in patients with systemic rheumatic disorders. Methotrexate exerts anti-inflammatory effects in adipose tissue and reduces cardiac fibrosis following experimental cardiac injury.113 Its use in rheumatic disorders reduces pro-inflammatory cytokines and has been associated with a lower risk of HF hospitalisations.114
The IL-1 family has been implicated in adipose tissue inflammation, and experimental inhibition has been accompanied by decreases in pro-inflammatory cytokines and cardiac fibrosis. In a small cross-over trial, the IL-1 antagonist anakinra reduced systemic inflammation and improved exercise capacity in patients with HFpEF.115 More impressively, in a randomised controlled trial of >10,000 patients with evidence for systemic inflammation, long-term treatment with the IL-1beta antagonist canakinumab decreased inflammatory biomarkers and the risk of hospitalisation for HF.116
The use of antagonists of TNF-alpha in the management of patients with rheumatic diseases (who are prone to HFpEF) has been associated with amelioration of myocardial inflammation, coronary microvascular dysfunction and cardiac functional abnormalities, and little change in or a reduced risk of HF events.117–121 These findings stand in contrast to concerns that the use of antagonists of TNF-alpha may exacerbate the clinical course of HFrEF (as is commonly seen following MI).122,123
If activation of the leptin-aldosterone-neprilysin axis plays a role in mediating the pro-inflammatory expansion of EAT,25 then drugs that modulate this axis could influence the course of HFpEF in patients who have rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis. Trials of mineralocorticoid receptor antagonists and neprilysin inhibitors in patients with HFpEF have not specifically targeted patients with systemic rheumatic disorders. However, it is noteworthy that these systemic rheumatic disorders are particularly common in women;124–127 and on subgroup analysis the benefits of mineralocorticoid receptor antagonists and neprilysin inhibitors have been reported to be greater in women than in men.74,128
Conclusion
Many systemic inflammatory rheumatic diseases, that is, rheumatoid arthritis, systemic lupus erythematosus, psoriatic arthritis and ankylosing spondylitis, are accompanied by an increased risk of HF, which begins at the onset of diagnosis and increases in proportion to the clinical severity and duration of the systemic inflammatory process. HF is not the result of accelerated CAD and ischaemic myocardial injury, but instead is related to myocardial inflammation, coronary microvascular dysfunction and fibrosis leading to HFpEF. A conceptual framework is proposed to explain the evolution of HF in these patients. Pro-inflammatory mediators characteristic of systemic inflammatory states (e.g. leptin, aldosterone and neprilysin) can cause expansion and biological transformation of EAT, leading to coronary microvascular injury and fibrosis of the underlying myocardium. EAT volume is increased in these systemic rheumatic disorders, and 30–50% of afflicted patients have subclinical evidence of cardiac inflammation, microcirculatory dysfunction and fibrosis, as well as the structural and functional features of HFpEF. Current anti-inflammatory agents that are used for the treatment of these systemic rheumatic diseases have the potential to minimise the development of cardiac involvement and thereby reduce the risk of HF. In addition, it is possible that drugs that modulate the leptin-aldosterone-neprilysin axis could modify the cardiovascular consequences of systemic inflammation and change the clinical course of HFpEF in these patients.