







In the past decades, takotsubo cardiomyopathy (TTC) has emerged as a specific form of acute and transient myocardial disease, predominantly affecting postmenopausal women in the clinical setting.1–4 In general, myocardial involvement in this setting appears to harbour a regional pattern particularly extending beyond the territory of a single coronary artery (and usually without concomitant significant coronary artery disease [CAD]).2,4,5 Mechanistically, sympathetic hyperstimulation attributable to a variety of external and internal stressors has been regarded as the fundemental trigger of TTC evolution.3,5–8 Despite the lack of consensus on the definitive diagnosis of TTC, a variety of diagnostic criteria (e.g. Mayo and Gothenburg) have been previously proposed.1,8,9 Among these, the Gothenburg criteria suggest a variety of diagnostic hallmarks, including reversible wall -motion abnormalities, frequently in response to a stressful trigger; absence of other potential causes associated with these abnormalities (e.g. ischaemia and tachycardia); and a disproportionately low elevation of cardiac troponin (i.e. no or mild elevation).8,9 More recently, the European HF Association has also suggested additional clinical findings, including ECG changes and elevation of natriuretic peptides, for the diagnosis of TTC, along with recommending that hyperadrenergic conditions (including pheochromocytoma) no longer be considered as exclusion criteria.1
In general, acute coronary syndromes (ACS) and TTC appear to have a close analogy based on their clinical characteristics, including symptomatology and ECG, which suggests the urgent need for coronary angiogram to reach a final diagnosis.3,10 On the other hand, since TTC primarily emerges as a myocardial disease with an acute onset, it may also present with HF symptoms and signs, instead or on top of ACS-like admission characteristics. Therefore, this phenomenon should also be differentiated from a variety of acute HF syndromes, including myocarditis, that might even be more challenging in clinical practice.1,3 Acute HF in patients with TTC might, to a large extent, be attributable to left ventricular (LV) dysfunction, characterised by systolic and/or diastolic dysfunction, along with certain specific complications including LV outflow tract (LVOT) obstruction arising in various degrees and durations. Accordingly, the present paper aims to highlight the morphological and mechanistic characteristics of LV involvement in the setting of TTC, with a particular emphasis on clinical aspects of LV dysfunction in this setting.
Takotsubo Cardiomyopathy-associated Wall-motion Abnormalities
Morphological Types
In clinical practice, TTC generally presents with a variety of regional and transient cardiac wall -motion abnormalities (mostly akinesia and rarely hypokinesia) that may, in certain settings, elicit a significant decline in cardiac function.8,9 Among these, LV apical akinesia along with a basal hyperkinesia pattern has been described as the classical and most common variant (comprising about 50–80 % of cases) , primarily characterised by an apical ballooning appearance on LV imaging.1,8,9 However, other less common variants involving LV basal (inverted TTC), midventricular and lateral segments have also been increasingly reported.1,9 Moreover, TTC may present with biventricular or isolated right ventricular (RV) involvement in about one-third of cases , and may even emerge as a global or focal LV dysfunction. Interestingly, a gradual upward trend in the incidences of these atypical variants has been reported.9
Importantly, myocardial involvement in this setting usually appears to be circumferential and extends beyond the territory of a single coronary artery, demonstrating a sharp transition to the normal or hyperkinetic territories, possibly, in part, due to the anatomical distribution and varying involvement of cardiac sympathetic branches.1,24 These wall-motion abnormalities, along with potential complications (LVOT obstruction, etc.), are generally diagnosed on invasive LV angiogram or transthoracic echocardiogram (TTE). On the other hand, cardiac MRI using late gadolinium enhancement may be more valuable in certain settings, including RV involvement and, particularly, formefruste manifestations that might mimic acute myocarditis or ACS.1,3 These techniques are also of utmost value to monitor gradual recovery of wall-motion abnormalities.1 Regardless of the location, these wall-motion abnormalities generally normalise within hours to weeks, largely depending on the clinical severity of TTC attack, and a complete recovery at 12 weeks is highly likely.1,11
Mechanistic and Molecular Basis
Several theories have been suggested regarding the pathogenesis of transient wall-motion abnormalities in the setting of TTC. Systemic and/or local catecholamine surge along with a regional heterogeneity in cathecholamine sensitivity is the most popular and extensively investigated theory.1,8,12 Evolution of similar wall-motion abnormalities in response to iatrogenic and hormonal triggers associated with sympathetic hyperstimulation also substantiate this theory.13,14 On the other hand, the exact mechanisms underlying excess cathecholamine toxicity in this setting have yet remained to be established. One such mechanism might be attributable to the supply–demand mismatch in oxygen and adenosine triphosphate (ATP) levels potentially leading to a state of metabolic shutdown, along with a significant decline in contractile functions at the myocellular level.8 Moreover, adrenergic hypersensitivity (and hence detrimental effects) may also appear to be potentiated in the setting of enhanced myocardial wall stress associated with sympathetic hyperstimulation or other causes.8 Within this context, even though the theories of macro- and microvascular dysfunction generally fail to fully explain the evolution of specific wall-motion abnormalities in the setting of TTC,15,16 myocardial ischaemia may potentially have a facilitating impact, and may decrease the threshold for the emergence of contractile dysfunction owing to impaired myocardial metabolism and energetics in this setting.8 Similarly, oestrogen deficiency may also be proposed, at least, to have a contributory role, largely through induction of endothelial dysfunction, for example.5
Excess cathecholamine toxicity along with severe energy depletion (and hence metabolic shutdown) also appear to be associated with a variety of corresponding histopathological changes at the myocellular level. Accordingly, myocardial biopsies obtained from acute TTC cases were previously found to have large areas of intracytoplasmic glycogen; damaged mitochondria (indicative of energy deprivation); quantitative and qualitative derangement of myocyte proteins (actin, titin, etc.); extracellular matrix enlargement harbouring interstitial fibrosis (increased collagen-1 and fibronectin); and myocyte disarray (as shown through connexin-43 labelling); however, there were no signs of cellular necrosis or apoptosis.17 These abnormal changes associated with myocardial dysfunction (both systolic and diastolic) were also shown to significantly normalise in the recovery phase of TTC.
On the other hand, altered LV geometry associated with enhanced myocardial wall stress might exclusively lead to the emergence of characteristic wall-motion abnormalities, suggesting a purely mechanical basis of TTC in certain settings.6,18–20 Circumferential wall stress (CWS) within the myocardium is well known to be associated with intraventricular pressure, chamber diameter and wall thickness.18,20 Accordingly, LV geometric changes, including septal hypertrophy and small LV cavity (usually due to hypertrophic cardiomyopathy or hypertensive heart disease) might potentially account for acute and substantial increments in intraventricular gradient, eventually giving rise to the evolution of apical ballooning (largely due to enhanced apical intraventricular pressure and hence CWS).18,19 Besides the substantial impact of intraventricular pressure gradient, relative enhancement of CWS in the apical in comparison to the basal segments might also be ascribed to the minor alterations in chamber diameters and wall thicknesses of the apical segments.20 Importantly, CWS increase in the apical segments may be persistent in nature and, as mentioned previously, may potentially create a milieu for adrenergic hypersensitivity within these segments. More interestingly, a recent study using cardiac MRI to measure LV wall stress in patients with TTC suggested that elderly women might have more pronounced end-systolic and end-diastolic wall stresses in response to hypertension due to stressful triggers, which may partly explain the specific age and gender predilection of TTC.21
Lastly, the beta-2 adrenoceptor/inhibitory G protein activation theory was recently suggested to specifically explain the evolution of apical akinesia due to TTC.8,22 According to this theory, excessive levels of adrenaline (but not noradrenaline) in the setting of acute stress potentially hyperstimulate beta-2 adrenoreceptors predominantly localised in the apical regions, which, in turn, activates the inhibitory G protein leading to a reduction in cyclic adenosine monophosphate levels, and hence contractile dysfunction, in these regions.8,22 However, the theory fails to explain the genesis of wall-motion abnormalities in the regions other than the apices, and it needs to be tested through further studies.
In summary, the pathogenesis of transient wall -motion abnormalities in the setting of TTC appears to be a quite unique process with a complex and multifaceted nature, with multiple factors implicated (adrenergic surge, changes in adrenoceptor sensitivity and enhanced myocardial wall stress, etc.), usually in various combinations. Importantly, these wall -motion abnormalities might give rise to the evolution of acute HF characterised by significant systolic and/or diastolic dysfunction in a portion TTC cases.
Left Ventricular Dysfunction and Clinical Heart Failure: An Ominous Duo in Patients with Takotsubo Cardiomyopathy
In general, it might not be as rare to encounter significant LV dysfunction and acute HF in patients with TTC as was considered previously. A previous retrospective analysis reported an HF incidence of 45 % on admission among patients with TTC.2 Moreover, 20 % of HF cases also suffered cardiogenic shock in this study. It is noteworthy that TTC, in contrast to the consensus on its favourable nature,23 may be associated with acute systolic and/or diastolic dysfunction on admission or during hospital stay, indicating particular management strategies.1 As described later in this article, the combination of LV dysfunction and clinical HF seems to have a much worse prognosis than subclinical LV dysfunction in the acute setting. On the other hand, the Gothenburg criteria suggest the presence of normal or near-normal cardiac output and LV filling pressures as supportive findings for the diagnosis of TTC.8 This potentially implies that HF associated with LV dysfunction should not be regarded as a routine finding in patients with TTC. Therefore, alternative causes of acute HF should also be considered beforehand in the clinical setting.

Systolic Dysfunction: Potential Causes and Implications
In the setting of TTC, acute HF due to LV systolic dysfunction, as determined by low LV ejection fraction (LVEF) values (below normal limits on TTE or other imaging modalities, including LV angiogram, MRI, etc.) has been regarded as the most common complication (in 12–45 % of cases) that warrants urgent diagnosis and proper management.1,2 Since LV systolic dysfunction in patients with TTC mostly evolves acutely, without pulmonary or systemic adaptive mechanisms, it generally presents with signs and symptoms of clinical HF, including dyspnoea, hypotension, pulmonary rales, etc.2 Importantly, there is also a strong correlation between the degree of acute systolic dysfunction and the severity of HF symptoms in a significant portion of TTC cases (as opposed to the setting of chronic cardiomyopathy and its symptomatology). On the other hand, cases with borderline systolic dysfunction ( LVEF >40–45 %) may remain clinically silent in terms of HF symptomatology. Conversely, it seems more reasonable to consider clinical HF as a highly probable scenario rather than an unpredictable complication in TTC patients with lower LVEF values (LVEF <40 %).2 Within this context, a retrospective analysis suggested that low LVEF (<40 %), presence of a physical stressor and advanced age (>70 years) as independent predictors of clinical HF in patients with TTC.2 However, this study grossly focused on the overall evolution of clinical HF that might also have risen because of the acute diastolic dysfunction.
Mechanistically, it seems likely that LV systolic dysfunction in patients with TTC might be attributable to a variety of factors, including extensive TTC-induced wall-motion abnormalities; decreased compensatory hypercontractility of the residual myocardial segments (due to bystander CAD,1 etc.); pre-existing myocardial disease;11 concomitant acute conditions (including ACSs)10 and mechanical complications.1,11 In general, evolution of systolic dysfunction is largely attributable to the direct impact of acutely evolving and extensive wall-motion abnormalities in TTC cases with previously normal or near-normal cardiac function. However, a portion of cases might already harbour a pre-existing myocardial disease that might clinically worsen because of a superimposing TTC. In other terms, a TTC attack might potentially account for an acute exacerbation of a chronic myocardial disease. On the other hand, it may be quite challenging to diagnose a new-onset TTC in these patients who already have segmentary or global wall-motion abnormalities.24
Traditionally, TTC and ACS appear to have a significant intersecting zone in terms of their clinical findings.3 Nevertheless, they should not always be regarded as mutually exclusive entities , even if coronary imaging suggests ACS/TTC co-existence in certain settings.10 A variety of clinical factors, including severe systemic inflammation on admission, an existing physical stressor and presence of spontaneous coronary artery dissection on coronary imaging, were recently suggested to predict this co-existence in clinical practice.10 Therefore, a co-existing ACS might contribute to, or even account for, the evolution of systolic dysfunction in certain TTC patients. Similarly, co-existing myocarditis, as the primary or secondary pathology, might also account (though much more rarely) for the significant systolic dysfunction in certain patients.3 As expected, LV systolic function in TTC patients with a co-existing ACS or chronic myocardial disease (e.g. cardiomyopathy), as opposed to those with TTC in isolation, does not completely normalise even after full recovery of TTC.
Lastly, mechanical complications, including acute LVOT obstruction and acute mitral regurgitation (mostly due to systolic anterior motion or apical tethering of subvalvular structures), along with very rarely encountered myocardial perforations, such as ventricular septal rupture (<1 % of cases),1,11,25–27 may also be regarded as specific forms of systolic dysfunction in patients with TTC. Potential causes of systolic dysfunction in the setting of TTC are summarised in Table 1. Among the mechanical complications, acute LVOT gradient merits further mention.
Acute Left Ventricular Outflow Tract Obstruction
Acute LVOT gradient (generally ranging between 20 and 140 mm Hg) has been considered a specific form of LV dysfunction generally encountered in up to 25 % of patients with TTC.1,11,26 In other terms, it may potentially be labelled as a form of systolic HF when associated with a reduction in cardiac output along with haemodynamic compromise. LVOT gradient in this setting is largely associated with the hypercontraction pattern of basal LV segments usually characteristic of the typical TTC variant, and is usually accompanied by systolic anterior motion and mitral valve regurgitation.1,25 Accordingly, a recent study demonstrated typical TTC incidences of 100 % and 78 % in TTC patients with and without an LVOT gradient, respectively.25 More importantly, the incidence of clinical HF was strikingly higher in TTC patients with a LVOT gradient as compared with those without in this study (78 % versus 28 %). In general, a LVOT gradient of >25 mmHg is regarded to have haemodynamic significance, and a value of ≥40 mmHg poses a high risk in clinical practice.1
On the other hand, the issue of whether LVOT gradient in the setting of TTC appears to serve as the cause or consequence of characteristic wall-motion abnormalities remains controversial.11,26 Even though mechanical factors (e.g. acute increases in intraventricular gradient) per se might serve as the fundamental contributor to the evolution of wall -motion abnormalities in certain settings,18,19 it seems more likely that LVOT gradient is a fully reversible complication usually arising during the early course of TTC.11,25,27 More interestingly, LVOT gradient might rarely emerge as a late-onset phenomenon with a persistent or permanent nature in the setting of TTC as well.27–29 Mechanistically, evolution of this phenomenon was previously ascribed to the TTC-induced temporal changes in LV geometry in the setting of a background myocardial disease, including hypertrophic cardiomyopathy.27 However, late-onset LVOT gradient, as opposed to its early onset counterpart, generally presents with classical symptoms of aortic stenosis (angina, dyspnea, etc.) rather than acute haemodynamic compromise.
Diastolic Dysfunction: The Forgotten Tale in Takotsubo Cardiomyopathy
In clinical practice, clinicians rarely focus on potential mechanisms, diagnostic algorithms or the clinical relevance of diastolic dysfunction in the setting of TTC. This attitude is largely based on the general misconception that considers existing diastolic dysfunction in this setting only as an innocent bystander, rather than a clinical phenomenon with important implications. In general, acute diastolic dysfunction arises in a significant portion of cases with TTC in various degrees and durations.6,30–33 Accordingly, in a recent study primarily focusing on the long-term prognostic impact of LV dysfunction in patients with TTC, the prevalence of diastolic dysfunction on admission was reported to be as high as 53 % (108 of 205 cases).31 Among these, the majority of patients (65 of 108) were found to have a mild degree of diastolic dysfunction (mitral E/A ratio <0.8, E/e′ ratio < 8, deceleration time >200 ms and normal left atrial [LA] filling pressure [grade I]34), with only a minority (13 of 108) having severe diastolic dysfunction (E/A ratio ≥2, increased LA filling pressure and deceleration time <160 ms [grade III]). In contrast with an asymptomatic nature of mild diastolic dysfunction at rest,34 moderate and severe degrees (grade II and III; pseudonormalisation and restrictive pattern on TTE, respectively) are more likely to elicit symptoms and signs of diastolic HF even at rest, and particularly in a more pronounced manner when they evolve acutely, as in the setting of TTC. Temporally, diastolic dysfunction arising after a TTC attack may be transient, persistent or even permanent.
Transient Diastolic Dysfunction
In the setting of TTC, this form of diastolic dysfunction is generally considered to arise in close temporal correlation with reversible segmentary wall-motion abnormalities in terms of its time of onset and duration.33 However, clear-cut mechanisms of this phenomenon remain to be fully established. At the myocellular level, both contraction and relaxation largely mediate their effects through a variety of common molecular targets, including interaction of myofilaments that are primarily governed by an energy-dependent, active process.31,35 Consistent with this, any severe metabolic shutdown of diverse aetiology at the cellular level (excess cathecholaminergic toxicity, etc.)8 is expected to elicit myocardial stunning or severe myocardial failure, substantially compromising both systolic and diastolic function in a simultaneous manner. In TTC hearts with full-blown myocardial stunning rather than hypokinesia in the affected region, the particular phase of cardiac cycle in which the myocardial stunning takes place is also of crucial importance in terms of emergence as well as severity of acute diastolic dysfunction. For instance, myocardial stunning at the mid- or end-systolic phase (frozen in a “neither contracting nor relaxed” state) is more likely to elicit significant diastolic dysfunction as compared with that occurring in the end-diastolic phase.
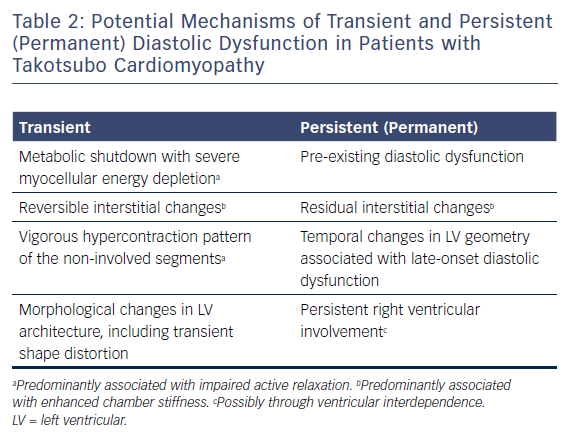
Interestingly, diastolic dysfunction might be associated not only with impaired active relaxation but also with enhanced passive LV stiffness in patients with TTC. Accordingly, LV diastolic stiffness was shown to be significantly higher, along with reduced atrial contribution to LV volume, during late diastole in a population of 24 TTC patients compared with a control group,36 possibly as a result of TTC-induced histopathological changes such as interstitial fibrosis,16 along with morphological changes in LV architecture, including shape distortion. Of note, non-involved segments might also contribute to diastolic dysfunction, possibly due to the hypercontraction pattern within these segments potentially leading to reduced relaxation, particularly in the presence of increased heart rate. Regardless of putative mechanisms, clinicians mostly consider diastolic dysfunction in this setting as a transient phenomenon; accordingly, a recent study has demonstrated complete recovery of diastolic function (as measured with conventional and tissue Doppler indices) in close correlation with systolic function in a population of 28 patients with TTC.33
Persistent or Permanent Diastolic Dysfunction: An Intricate and Multifaceted Phenomenon
It seems quite surprising that TTC, a phenomenon universally renowned for its apparently fully reversible nature, might be associated with persistent (or even permanent) diastolic dysfunction that potentially extends far beyond the recovery of its characteristic wall -motion abnormalities. Within this context, a previous study, in which cardiac MRI was used to evaluate serial changes in systolic and diastolic function in TTC patients, clearly demonstrated the relative persistence of diastolic dysfunction (as measured with indices including LV peak filling time and LA emptying volume) in comparison to systolic dysfunction (as measured with LVEF) at the time of discharge.32 However, both diastolic and systolic function were reported to fully recover on follow-up. This implies that diastolic dysfunction, despite its ultimate normalisation, may tend to demonstrate a significantly slower and more delayed recovery pattern compared with systolic dysfunction in patients with TTC. This notion is in line with a recent study that demonstrated improvement of diastolic function in only 28 % of cases in the recovery phase (mean 38±16 days after admission) , with the rest (72 %) having unchanged or worsening diastolic function.31 Importantly, persistent diastolic dysfunction in isolation might also be regarded as a trigger of acute HF, and hence not just an innocent bystander in TTC patients. Accordingly, a case of acute HF (arising 1 month after recovery from a TTC attack) due to persistent diastolic dysfunction was previously reported as the first case in the literature,37 suggesting the particular clinical relevance of this phenomenon.
Mechanistically, persistent or permanent diastolic dysfunction seems to be a more complex and multifaceted phenomenon compared with its transient counterpart.38,39 Microstructural changes, including enlargement of the extracellular matrix and interstitial fibrosis,17,40 which are generally considered transient, might, to some degree, outlast the acute stage of TTC, and might even persist indefinitely.38,39 Besides these residual microstructural changes,39 persistent RV involvement might also have a pivotal role in the evolution of persistent or permanent diastolic dysfunction, largely through ventricular interdependence and TTC-induced temporal changes in cardiac macrostructure, including LV geometry.6,38 However, pre-existing diastolic dysfunction seems to be the most likely aetiology given the advanced age of the TTC population.31,38
Table 2 demonstrates the potential mechanisms of transient and persistent (permanent) diastolic dysfunction in patients with TTC.
Management of Left Ventricular Dysfunction
Since LV dysfunction in this setting is regarded as a fully reversible phenomenon (at least to systolic function at rest), supportive measures have been the mainstay of therapy in clinical practice.1,11 Renin–angiotensin system (RAS) blockers, beta-blockers (preferably metoprolol and carvedilol based on preclinical studies)41,42 have been generally recommended in the acute setting.1,11 On the other hand, the recent European HF Association position statement recommended their use exclusively in patients with a LVEF of < 45 %.1 However, in case of an emerging clinical systolic HF, these agents (regardless of initial LVEF values) should be initiated, at least until recovery. Diuretics and nitroglycerin may also be recommended , largely for symptomatic relief, after exclusion of LVOT obstruction.11
Severe forms of acute HF (significant pulmonary oedema, hypotension, etc.), with or without cardiogenic shock due to primary pump failure, generally mandate the use of mechanical support devices such as extracorporeal membrane oxygenation and LV assist devices, all serving as bridge-to-recovery strategies.11,43 On the other hand, the use of intra-aortic balloon counterpulsation (IABP) in this setting is controversial, and is not routinely recommended based on its neutral results in recent trials.1 Importantly, cathecholaminergic inotropes (dobutamine, isoprenaline, adrenaline, etc.) are strictly contraindicated because they do more harm than good and might worsen the clinical status in this setting, largely due to their paradoxical adverse impact on myocardial function.1,44 On the other hand, non-cathecholaminergic inotropes, including levosimendan, might serve as a safer and efficient alternative to manage severe HF in this setting.45,46
In the setting of acute significant LVOT obstruction (with a gradient of ≥40 mmHg along with a systolic blood pressure of <110 mmHg), the initiation of selective alpha-1 agonists, including phenylephrine and beta-blockers (including short-acting intravenous ones), along with the use of intravenous fluids, may be the preferred strategy.1,11 On the other hand, an existing severe pulmonary oedema and/or cardiogenic shock should initially warrant the urgent use of mechanical support until the mitigation or recovery of LVOT obstruction. Importantly, all inotropic agents and IABP are generally considered to be contraindicated because of their significant potential to aggravate the gradient.11 Therefore, even before initiating levosimendan (the only indicated inotrope for the management of severe primary pump failure in TTC patients)45,46 an existing significant LVOT gradient (accompanying or accounting for the severe clinical status) should be definitively ruled out beforehand.
Currently, there exists no ideal therapy for the management of diastolic dysfunction; accordingly, many agents that work well in subjects with systolic HF generally fail or do not work efficiently in those with diastolic HF, particularly in terms of hospitalisation and mortality rates.47 This notion might, to a large extent, apply to the setting of TTC as well. Symptomatic relief (diuretics, etc.) along with, where possible, rate control with beta-blockers, for example, (and hence enhancement of LV diastolic filling) may be the fundamental strategy in TTC cases with diastolic HF in isolation or in combination with systolic HF. In general, severe diastolic dysfunction (with a restrictive pattern) with or without life-threatening symptomatology (pulmonary oedema, haemodynamic compromise) appears to be a quite rare phenomenon.31 In the long term, agents with proven mortality benefit, including nebivolol, may be specifically preferred, particularly in elderly TTC patients with persistent or permanent diastolic dysfunction.48
Left Ventricular Dysfunction as a Risk Stratifier: Shor- and Long-term Impacts on Prognosis
In the recent European HF Association position statement, a variety of clinical parameters , including advanced age (≥75 years ), an LVEF value of <35 %, pulmonary oedema, an LVOT gradient of >40 mmHg, systolic blood pressure of <110 mmHg and other life-threatening mechanical or arrhythmogenic complications, have been suggested as the major high-risk factors in the acute setting of TTC.1 This potentially denotes that LV functions and associated haemodynamic indices might be regarded as the most crucial determinants of short-term prognosis, possibly associated with the majority of adverse events, including in-hospital deaths among patients with TTC. However, the prognostic impact of clinical LV dysfunction (in combination with symptoms and signs) seems to be more substantial compared with subclinical LV dysfunction in the acute phase.
On the other hand, little is known about the long-term prognostic impact of transient LV dysfunction in this setting.1 It is well known that adverse myocardial remodelling (characterised by late-onset and gradual changes including compensatory hypertrophy, ultimately leading to a progressive and insidious decline in systolic function)49 generally evolves in survivors of ACS, myocarditis, etc., with significant myocardial injury, and appears to be associated with poor prognosis in these patients. However, such a limited duration of systolic dysfunction, as in the setting of TTC, can not be considered a trigger of adverse myocardial remodelling, which generally evolves and progresses within months to years after the initial insult.
In contrast, LV dysfunction extending beyond the acute stage might have long-term consequences in the setting of TTC; accordingly, persistence of diastolic along with systolic dysfunction (LVEF <50 %) in the recovery stage of TTC (38±16 days) was suggested to be associated with both cardiac and non-cardiac adverse events over a median period of 2 years in a retrospective analysis.31 These authors primarily suggested pre-existing diastolic dysfunction and associated patient-related factors, such as frailty, as the potential mechanisms of long-term adverse events. In particular, cardiovascular events (including arrhythmogenesis) in the long term may be associated, to some degree, with persistent myocardial changes, including interstitial fibrosis, in TTC patients, mostly arising as part of an ongoing clinical LV dysfunction.6,31 Within this context, use of RAS blockers (up to 1 year after recovery) was previously suggested to improve prognosis, largely owing to the favourable impact of these agents on myocardial microstructure.11 However, potential TTC recurrences (in 5–22 % of cases within 5 years), as well as comorbidities including malignancy,1,50 have been generally considered as the fundamental determinants of long-term prognosis in TTC patients.
Importantly, frequent TTC recurrences might theoretically be associated with cumulative pathological changes within the myocardium, even leading to adverse remodelling in rare instances. Within this context, beta-blockers and sympathetic ganglion blockade, as well as psychological (and cognitive behavioural) treatment and certain yoga practices, may improve long-term prognosis in selected cases, possibly through prevention of future TTC recurrence and, correspondingly, abortion of cumulative myocardial changes, to some degree.1,27,51–53 These preventative implications appear to be based on the strong association between acute autonomic discharge and TTC, and may also be substantiated by the recent evidence of a lower prevalence of diabetes mellitus among TTC cases, potentially suggesting the protective role of diabetes-induced sympathetic neuropathy against TTC evolution.51–53
Subclinical Left Ventricular Dysfunction: A Subtle Phenomenon with Significant Impact on Quality of Life Among Takotsubo Cardiomyopathy Survivors
A portion of cases remote from the index TTC event may continue to suffer a variety of symptoms, including reduced exercise capacity, palpitation and angina, even with fully recovered systolic and diastolic function, as measured with conventional TTE indices.1,54 On the other hand, these patients were generally found to have abnormal tissue Doppler parameters, including impaired myocardial deformation indices (LV global, circumferential and longititunal strain) compared with controls.54,55 In a recent, interesting study of 37 cases with a TTC history and 37 control subjects, a significant portion of cases (88 %) were reported to have symptoms of HF after a median duration of 20 months after the index TTC attack, including fatigue, breathlessness and angina, despite normal LVEF values on TTE.54 Accordingly, these patients, compared with controls, had significantly impaired exercise capacity on exercise testing (decreased peak levels of O2 consumption: 24±1.3 vs 31±1.3 ml/kg/min, p<0.001), as well as reduced global longitutinal (–17±1 vs –20±1 %, p=0.006) and apical circumferential strain rates (–16±1.0 vs –23±1.5 %, p<0.001). Moreover, these patients were also found to have impaired myocardial energetic status, as demonstrated by a reduced phosphocreatine/ATP ratio (1.3±0.1 vs 1.9±0.1, p<0.001) compared with control subjects.
These findings may imply that even though macroscopic normalisation, as demonstrated with conventional TTE indices, generally appears to be obvious, recovery at the myocellular level may not be complete, with persistence of dysfunction, to some degree, in cellular components (mitochondria, contractile proteins, etc.) probably on a parallel with the clinical severity of the index event in a significant portion of TTC survivors. In other terms, some sort of myocardial dysfunction (systolic and/or diastolic) arising during cycles of enhanced myocardial workload (exercise, hypertensive attacks, etc.), largely because of the residual dysfunction in myocardial energy metabolism and contractile reserve, may account for this poorly understood phenomenon. Myocardial failure on exercise may also be verified with the use of certain tools, including diastolic stress testing56 and dobutamine stress echocardiogram, in these patients.
Beta-blockers may be of particular benefit for symptomatic improvement in this setting.1 In addition, agents such as trimetazidine and ranolazine, which primarily target improvement of cellular energy metabolism, and hence myocardial ischaemia, were recently suggested as promising adjunctive options for the management of HF.57 Accordingly, TTC survivors with persistent symptomatology may be the ideal candidates for such therapeutic strategies in clinicalpractice. Importantly, since future TTC attacks might further aggravate sublinical LV dysfunction, prevention of TTC recurrences is also of paramount importance in this setting. Within this context, (besides the well-known effects of beta-blockers, psychological therapy, etc.), isoflurane (an anaesthetic agent) was recently found to be effective both in the prevention and attenuation of TTC-induced LV dysfunction in experimental studies.58,59 However, further studies are still warranted to discover novel agents with preventive features and disease-specific actions (targeted therapy) in the setting of TTC.
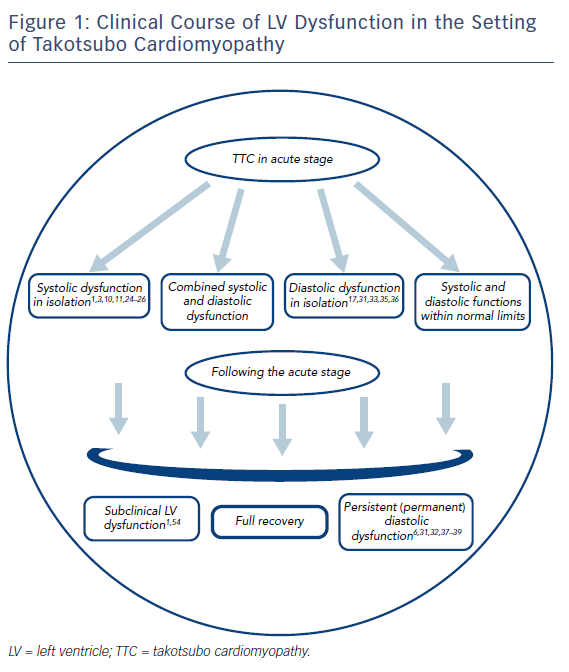
Figure 1 demonstrates the general clinical course of various patterns of LV dysfunction in patients with TTC.
Conclusion
In clinical practice, a significant proportion of patients with TTC present with LV dysfunction (with or without clinical HF) that may be regarded as an umbrella term, primarily constituting a variety of clinical patterns with diverse characteristics and implications in the short and long term. Accordingly, systolic dysfunction (defined as a resting LVEF below the normal reference values on imaging modalities) generally emerges as a fully reversible pathology, primarily serving as an important determinant of prognosis in the acute setting. Moreover, transient diastolic dysfunction, in isolation or in combination with systolic dysfunction, may also add to the symptomatology and, to some extent, prognosis of TTC in the acute setting. On the other hand, recent observations suggest that TTC, despite the gross recovery of LV function on LV imaging, may not always be considered a fully reversible phenomenon, as demonstrated by the persistence of certain pathologies, including diastolic dysfunction and subclinical LV dysfunction, in a significant proportion of cases. Moreover, these persistent and subtle changes may have the potential to significantly influence long-term prognosis, as well as quality of life in TTC survivors.
Unfortunately, there exists no specific targeted therapy for the management of acute and/or residual LV dysfunction in the setting of TTC, and the use of supportive measures is generally indicated as the fundamental strategy both in the short and long term. Therefore, future clinical studies should particularly focus on novel agents specifically targeting improvement of the molecular alterations underlying myocardial dysfunction in the setting of TTC. Disease-specific strategies, on top of supportive measures, might substantially speed up the normalisation of transient myocardial dysfunction, along with mitigating or aborting persistent myocardial changes, and hence might significantly improve overall prognosis.