










In randomised controlled trials (RCTs), treatment with β-blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and mineralocorticoid receptor antagonists reduce mortality and/or hospitalisation of patients with heart failure with reduced ejection fraction (HFrEF), but these various drugs have not similarly benefited those with heart failure with preserved ejection fraction (HFpEF). Recently, several RCTs have shown clinical benefit in HFpEF with sodium–glucose cotransporter 2 inhibitors (SGLT2is; Table 1), potentially being the first drug class to provide relief for this heterogenous and difficult to treat patient population. This is a critical review of the RCTs of SGLT2is in HFpEF, and the numerous SGLT2i proposed underlying mechanisms of action in HFpEF.
Randomised Controlled Trials of SGLT2 Inhibitors in Patients with HFpEF
Phase III Randomised Controlled Trials
In November 2020, the SOLOIST-WHF trial results were groundbreaking. For the first time, a RCT treatment effect was demonstrated with SGLT2is in HFpEF patients.1 A prespecified subgroup analysis suggested a significant decrease in the modified primary outcome of cardiovascular (CV) deaths, hospitalisation for heart failure (HHF) and urgent visits for heart failure (HF) in HFpEF patients with diabetes (HR 0.48; 95% CI [0.27–0.86]) receiving sotagliflozin versus placebo, and it was achieved in 28 days.1,2 SOLOIST-WHF was also the first to show initiation of SGLT2 inhibition in HFpEF patients with acute HF, in stabilised patients preceding discharge or shortly afterwards – resulting in a lower total number of CV deaths and HHF, and urgent visits for HF versus placebo.1 However, due to the small sample size of HFpEF patients (n=256) with ejection fraction (EF) ≥50% and an early cessation of the trial due to the COVID-19 pandemic, firm conclusions were cautioned against, and the completion of future RCTs, such as EMPEROR-Preserved, PRESERVED-HF, CHIEF-HF and DELIVER, were highly anticipated.1
In August 2021, the results of EMPEROR-Preserved were released. The RCT assigned 5,988 patients with class II–IV HF and an EF >40% to receive empagliflozin versus control, and demonstrated a significant reduction in the primary outcome (composite of CV death or HHF); however, that was mostly related to a 29% lower risk of hospitalisations (HR 0.79; 95% CI [0.69–0.90]; p<0.001).3 These protective effects were observed in patients with and without diabetes.3 Importantly, empagliflozin appeared to have less effect as left ventricular EF increased (EF ≥50%) – still demonstrating a significant reduction in first HHF, but without significant reductions in CV mortality or total HHF.3,4 Empagliflozin may be more beneficial for HFpEF patients with mildly reduced EFs (HFmrEF; EF 40–49%), because they have clinical features more similar to those of HFrEF; further analysis and future RCTs should clarify the effectiveness of SGLT2is in patients with ‘true’ HFpEF on the higher end of the EF spectrum (EF ≥50%).3–6
EMPEROR-Preserved also demonstrated a reduction in the incidence of outpatient visits for worsening HF, including the need for urgent care visits, reaching significance at 18 days and remaining significant throughout follow-up.7 In addition, empagliflozin was associated with a decreased risk of HHF requiring intensive care, or vasopressor or inotropic therapy, and a reduced need of increasing diuretic therapy in the outpatient setting.7 Within days of these significant EMPEROR-Preserved results, the Food and Drug Administration (FDA) granted Breakthrough Therapy designation to empagliflozin for the treatment of HFpEF, the first of the SGLT2is granted this status.8 Soon after, the FDA approved and expanded the HF indication for empagliflozin from HFrEF to also include HFmrEF and HFpEF.
In September 2021, PRESERVED-HF was the first trial to demonstrate SGLT2 inhibition (dapagliflozin) improves symptoms, physical limitations and 6-minute walking distance in patients with HFpEF.9 Functional status in patients with HFpEF improved in just 12 weeks regardless of diabetes status, and importantly, with a median EF of 60%.9 Of the 324 patients receiving dapagliflozin or placebo, the treatment group demonstrated an improvement in the Kansas City Cardiomyopathy Questionnaire (KCCQ)-Clinical Summary Score of 5.8 points (95% CI [2.3–9.2]; p=0.001), due to enhancements in both the KCCQ-Total Symptom Score of 5.8 points (95% CI [2.0–9.6]; p=0.003) and the physical limitations score of 5.3 points (95% CI [0.7–10.0]; p=0.026).9 Dapagliflozin also increased the 6-minute walk test by a mean of 20.1 m (95% CI [5.6–34.7]; p=0.007) and a KCCQ-Overall Summary Score of 4.5 points (95% CI [1.1–7.8]; p=0.009).9
Interestingly, the similar EMPERIAL-Preserved trial reported a non-significant 2.0-point increase in KCCQ-Total Symptom Score with empagliflozin versus placebo, and also did not demonstrate a significant change from baseline in the 6-minute walk test.9,10 The larger and significant functional status effect observed in PRESERVED-HF versus EMPERIAL-Preserved possibly has to do with the types of patients enrolled that had characteristics indicating substantial symptomatic and functional impairment at baseline (42 versus 22% with New York Heart Association class III/IV, and 244 versus 298 m in median 6-minute walk test, respectively).9,11 In addition, PRESERVED-HF versus EMPERIAL-Preserved included a higher percentage of patients necessitating loop diuretic therapy (88% versus 72%, respectively), and a higher amount with AF (53% versus 30%, respectively), which is associated with more right-sided HF and pulmonary hypertension with less cardiac reserve.12 Finally, PRESERVED-HF included larger proportions of women and African-Americans, with markedly higher BMI (34.7 versus 29.6, respectively), which is more representative of the US population.13
Further support for the impact SGLT2is may have on functional status in HFpEF was demonstrated in CHIEF-HF.14 Regardless of diabetes status, canagliflozin significantly improved symptoms and quality of life, as demonstrated by a KCCQ-Total Symptom Score of 4.5 points from baseline (95% CI [–0.3, 9.4]) for those with HFpEF (n=267; EF >40%) in as little as 2 weeks, and these improvements were sustained throughout the 3-month trial.14 However, as discussed earlier with EMPEROR-Preserved, data for CHIEF-HF included patients with HFmrEF (EF 40–49%) who appeared to be more similar to HFrEF in their clinical characteristics, and data may be less convincing in patients with EF ≥50.15
Finally, the highly anticipated DELIVER is a large RCT of 6,263 HFpEF (EF >40%) patients evaluating the effect of dapagliflozin versus control in reducing the composite of CV death, HHF and urgent HF visits, with an estimated completion date of March 2022.16 DELIVER should further clarify the possibility of SGLT2is reducing CV death in HFpEF patients.17 As stated earlier, uncertainty still remains regarding the effect of SGLT2is on mortality in HFpEF patients, as SOLOIST-WHF had a small sample size representing EF ≥50 (n=256), and EMPEROR-Preserved met its primary outcome mostly due to a reduction in the risk of hospitalisations, not mortality.1,3 DELIVER was also designed to recognise heterogeneity in the HF population at the higher end of the EF spectrum, and is powered with large sample sizes across the EF spectrum (41–49%, n=2,111; 50–59%, n=2,256; ≥60%, n=1,892).17 This should further elucidate the effect of SGLT2is in those at the higher end of the EF spectrum, where EMPEROR-Preserved data showed attenuation of benefit versus those with HFmrEF in reducing hospitalisations.3
Notably, DELIVER also included patients with recent acute decompensated HF requiring intravenous HF therapies or mechanical support, and should provide further data helping guide the role of SGLT2is in the inpatient and early post-discharge settings.17 More evidence for the use of SGLT2is in acute HF is emerging since the SOLOIST-WHF results (discussed earlier); post hoc and pre-specified analysis of the EMPULSE trial found that initiation of empagliflozin in patients hospitalised with acute HF produced clinical benefit regardless of EF.18 Results of the DELIVER trial will be available in 2022.17
Phase II Randomised Controlled Trials
Evaluation of the CAMEO-DAPA is a prospective, double-blind study (NCT04730947) with an estimated 46 participants and a primary outcome measuring the change in pulmonary capillary wedge pressure at baseline and 7 months during exercise. Patients included will have EF ≥50, BMI ≥30 and elevated pulmonary capillary wedge pressure (≥25 mmHg) during exercise at baseline. SGLT2is may reduce pulmonary capillary wedge pressure partly due to its unique diuretic ability (discussed in the next section) of decongesting fluid accumulation in the interstitial compartment without activation of the sympathetic nervous system.19,20 The trial is currently recruiting and has an estimated completion date of July 2023.
SGLT2i and KNO3 in HFpEF, the SAK HFpEF trial (NCT05138575) is a randomised crossover assignment trial in the recruiting stage with an estimated 53 participants, testing whether empagliflozin with and without potassium nitrate, improves submaximal exercise endurance, skeletal muscle oxidative phosphorylation capacity, intramuscular perfusion, and changes in the skeletal muscle metabolome, proteome and respiration in participants with HFpEF. Potential mechanisms regarding SGLT2i-induced metabolic changes in skeletal muscle and improvements in exercise capacity are discussed in the next section. The estimated completion date is September 2026.
The STADIA-HFpEF trial (NCT04475042) is in the recruiting stages, and is a randomised quadruple masked crossover study with a washout period, with an estimated 26 participants, investigating the effect of treatment with dapagliflozin on left ventricular distensibility in patients with early HFpEF. Proposed mechanisms of action of the effect of SGLT2is on cardiac stiffness are described in the next section. The estimated completion date is May 2022.
Proposed Mechanisms of Action of SGLT2is in HFpEF
SGLT2is Target SGLT2 and Na+/H+ Exchanger 3 in the Kidney
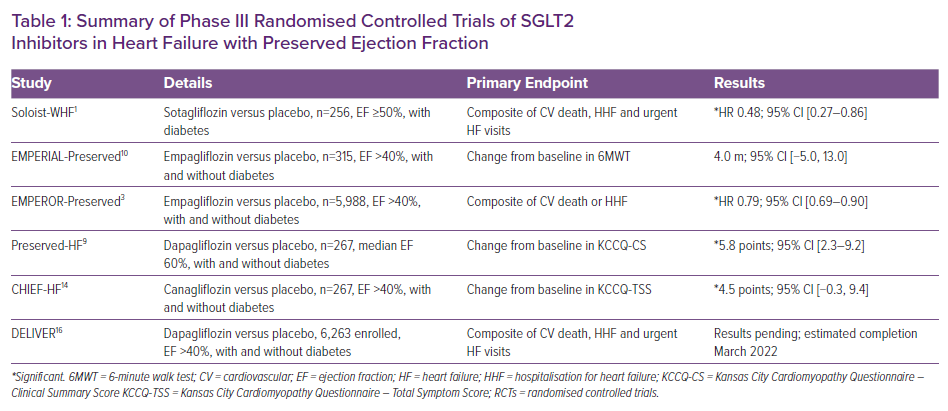
In the healthy kidney, most filtered glucose is reabsorbed in the proximal tubule by SGLT2, and most filtered sodium is reabsorbed by Na+/H+ exchanger 3 (NHE3).21–23 SGLT2 colocalises with NHE3, thereby SGLT2 inhibition also interferes with the function of NHE3.24 SGLT2is’ direct inhibition of SGLT2 and indirect inhibition of NHE3 induces glycosuria, osmotic diuresis, and natriuresis – mechanisms commonly proposed as providing potential benefit in HFpEF (Figure 1).7,24,25
The glucose-lowering and weight loss properties of SGLT2is alone cannot fully explain the CV benefits seen in HFpEF, since glycaemic control and other antihyperglycaemics that promote weight loss do not appear to decrease CV death or HF hospitalisations in HFpEF.26–28 The osmotic diuresis with resultant free water loss has been projected to explain the beneficial relatively higher interstitial versus intravascular fluid losses with a reduced reflexive neurohormonal activation thought to make SGLT2is unique when compared with conventional diuretics.20,29,30 SGLT2i-induced osmotic diuresis may improve myocardial oedema by increasing plasma oncotic pressure and stimulating plasma refill, thereby pulling fluid from the cardiac interstitium through Starling’s forces.25 A reduction in myocardial oedema is associated with a rightward shift in end-diastolic pressure volume relationship with decreases in left ventricular (LV) mass on cardiac magnetic resonance, and may be contributory in the early improvement of functional status demonstrated by PRESERVED-HF and CHIEF-HF, and may also partly explain EMPEROR-Preserved reaching significance in just 18 days with a reduction in worsening HF events.7,9,31 SGLT2is may also be a novel diuretic in that patient fluid response can vary depending on the baseline volume status of the patient, with less extracellular fluid loss in patients without fluid retention, potentially helping maintain a suitable body fluid status with lower risk of volume depletion.32
SGLT2i-induced natriuresis via inhibition of NHE3, which is upregulated in HF and implicated in diuretic resistance, may explain the reduced need for increasing outpatient diuretic therapy demonstrated by the EMPEROR-Preserved trial.7,33 Natriuresis may also be partly responsible for the reduced progression of kidney disease seen in people with diabetes, by increasing the sodium load to the macula densa with a resultant afferent arteriolar constriction and reduced glomerular hyperfiltration via tubuloglomerular feedback.34 Interestingly, SGLT2is (dapagliflozin) are also displaying prevention against the progression of chronic kidney disease in people without diabetes.35 Considering the bidirectional relationship of the heart and kidney, the kidney protection provided by SGLT2is may confer cardiac benefit indirectly, but this is speculative. In addition, long-term negative sodium balances associated with natriuresis have been shown to reduce aortic stiffness, potentially reducing the symptoms and progression of HFpEF.36,37
SGLT2i-induced natriuresis and diuresis may partly explain the reduced hospitalisations, and the easing of symptoms and physical limitations seen in HFpEF patients, but evidence suggests that SGLT2i mechanisms acting more directly on the heart may also play a significant role in their CV protection.7,9 SGLT2is activate sirtuins, reduce epicardial adipose tissue and its associated pro-inflammatory adipokines, promote cardiomyocyte sodium and calcium homeostasis, and relieve the right ventricle of pulmonary hypertension – all of which will now be discussed.24,38–41
SGLT2is Activate Sirtuins
Inhibiting SGLT2 in the proximal tubule produces glycosuria with calorie loss and a perceived state of starvation, thereby inducing a fasting-like state in tissues throughout the body, including the heart, skeletal muscle and kidney.24,42 Nutrient deprivation activates proteins called sirtuins (Sirt1, Sirt3, Sirt6), which are predominantly NAD+-dependent deacetylases involved in regulating autophagy, metabolism and mitochondrial function, thereby helping to alleviate cellular stress and inflammation.24,42–44 Autophagy, meaning self-eating, is cellular housekeeping to remove and/or recycle proteins and dysfunctional organelles – protecting the cells from oxidative stress, which is dysfunctional in the hearts of HFpEF patients.43–45 Increases in sirtuin activity are complex, but there may be a relationship between nutrient deprivation and a rise in NAD+ levels that activate sirtuins.42 Interestingly, HFpEF patients have reduced availability of NAD+, and impaired actions of Sirt1, Sirt3 and Sirt6.44 SGLT2i-induced increases in the abundance and activity of Sirt1, Sirt3 and Sirt6 in numerous tissues may explain some of the mechanisms behind their clinical benefits in HFpEF patients.40,44,46
Sirtuin 1
In cardiomyocytes, Sirt1 may induce autophagy directly by deacetylation of autophagy-related genes Atg5, Atg7 and Atg8, and may also increase autophagic flux by deacetylating the transcription factor forkhead box, class O isoform 1, increasing the expression of autophagy regulatory genes.47 The balance between the redox state and autophagy is crucial for cellular homeostasis, and inadequate basal autophagic flux and/or too many oxidants leads to a loss of redox balance that is related to the development of maladaptive cardiac remodelling.45 HFpEF pathophysiology includes damaged mitochondria with increases in reactive oxygen species (ROS), and also involves the downregulation of genes involved in autophagy.48,49 This cellular imbalance leads to oxidative damage, and promotes hallmarks of HFpEF: contractile dysfunction, cardiac stiffness, microvascular rarefaction, apoptosis, fibrosis and ECM remodelling.50 SGLT2i-induced calorie loss and resultant increases in autophagic flux through Sirt1 may mitigate HFpEF pathology by the clearance of dysfunctional mitochondria and alleviation of oxidative stress.40
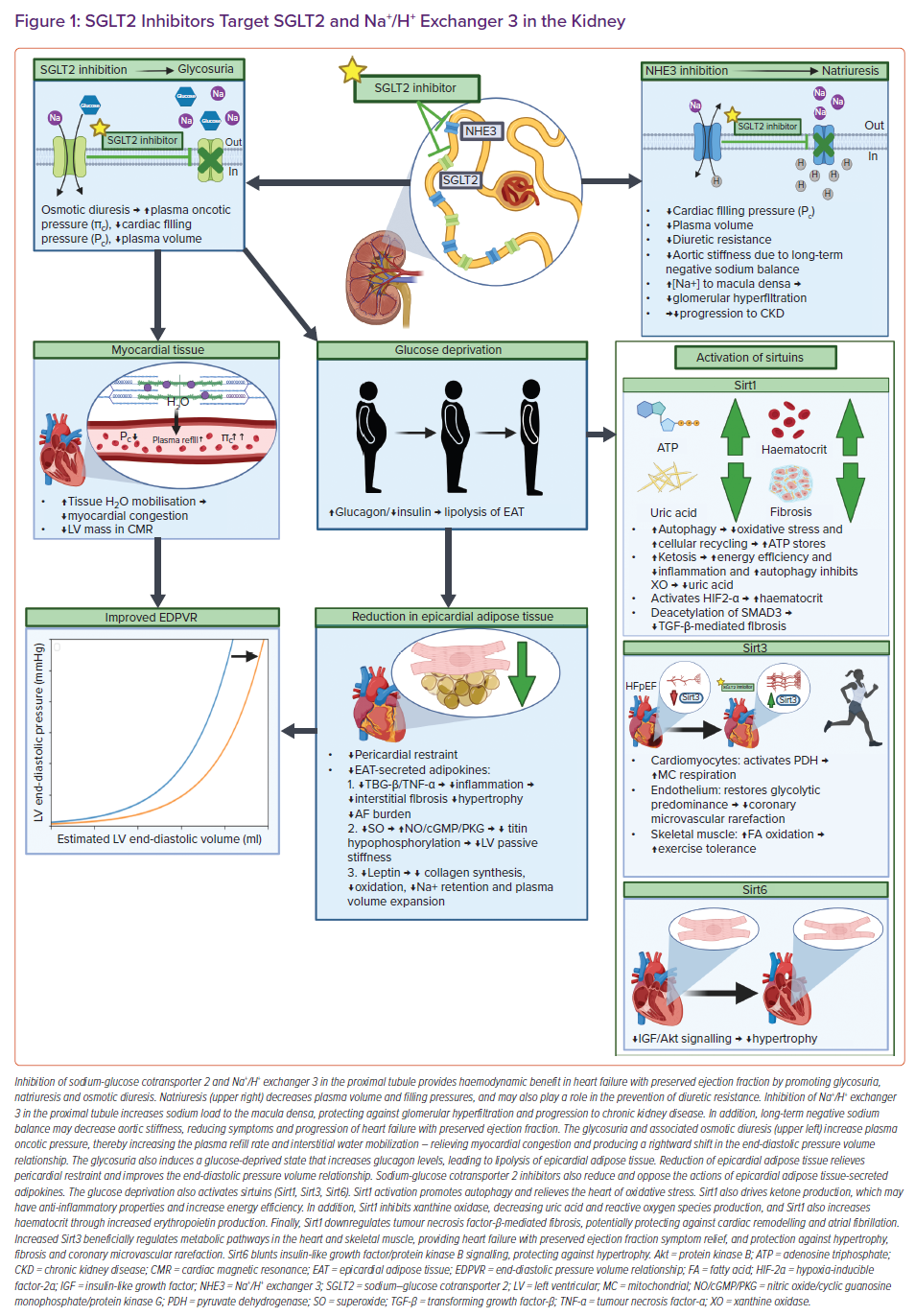
Sirt1 can also deacetylate and activate the transcriptional coactivator, peroxisome proliferator-activated receptor-γ coactivator 1-α.51–53 Like other Sirt1 interactions, this pathway may increase autophagic flux, but could also provide HFpEF patients benefit through the reprogramming of metabolism and driving ketosis through regulation of its rate-limiting step.52 The pro-inflammatory state induced by comorbidities in HFpEF is associated with increased ROS and cytokine production that overwhelm the anti-inflammatory defences of the myocardium and vasculature, contributing to cardiac fibrosis.54,55 Increased ROS in HF promotes depletion of anti-oxidants with mitochondrial DNA damage and a reduction in adenosine triphosphate (ATP) synthesis.56,57 It is hypothesised that SGLT2is induce mild persistent hyperketonaemia, specifically through the SIRT1/PCG-1α pathway, whereby b-hydroxybutyrate is oxidised in preference to fatty acids, with improvement of cardiac function and efficiency, although this is controversial.40,58,59 It does appear SGLT2is improve cardiac energy efficiency by recycling cellular constituents through autophagy and thereby increasing ATP stores.40 Interestingly, ketone bodies themselves may also have anti-inflammatory properties and stimulate autophagy.60,61 The contribution of ketone metabolism in HFpEF remains to be established, but SGLT2i mediated ketosis may improve cardiac energy efficiency, reduce pro-inflammatory cytokine-induced mitochondrial damage and protect against cardiac fibrosis in HFpEF patients.62
Another possible source of ROS in failing hearts is xanthine oxidase (XO), which is stimulated by oxidative stress.63,64 An important statistical determinant of CV benefit with SGLT2is is the lowering of uric acid, particularly in those with type 2 diabetes, which may apply to the cardiometabolic phenotype of HFpEF, although this is speculative.65–67 Notably, empagliflozin lowered uric acid levels, and consistently reduced CV outcomes in those with type 2 diabetes and CV disease versus placebo.67 SGLT2is may blunt XO activity by relieving oxidative stress. Furthermore, Sirt1 activation may also downregulate XO, which in turn, may beneficially upregulate PCG-1α protein expression.68,69 The complex relationship between XO, Sirt1 and peroxisome proliferator-activated receptor-γ coactivator 1-α could explain the uric acid-lowering effects of SGLT2is.40
Activation of Sirt1 through SGLT2i-induced nutrient deprivation signals also leads to deacetylation of hypoxia-inducible factor-2α, a transcription factor with the erythropoietin gene as its main target.66,70 This may be the mechanism responsible for increases in haematocrit seen clinically with SGLT2 inhibitors.71 An increase in red cell mass may improve myocardial oxygen delivery and help alleviate LV hypertrophy in HFpEF patients, and importantly, haematocrit is a significant statistical determinant associated with the reduction in adverse cardiac events seen with SGLT2is.65,72–75
Finally, in regard to Sirt1, Sirt1-mediated deacetylation of SMAD3 may provide HFpEF patients protection by downregulation of transforming growth factor-beta (TGF-β)-mediated fibrosis through inhibition of fibrotic transcriptional activity, and also by inhibiting myofibroblasts.44,76–79 Cardiac fibrosis plays an important role in HFpEF pathology, and contributes to cardiomyocyte stiffness and cardiac remodelling.80 In addition, fibrosis is linked to AF, which occurs in up to two-thirds of HFpEF patients prior to, concurrent with or subsequent to HF diagnosis, and is associated with higher mortality rates.81,82
Sirtuin 3
HFpEF is also associated with disrupted activity of Sirt3, a mitochondrial deacetylase with tissue-specific roles that is expressed in highly metabolically active tissues, such as the heart.44,83 Sirt3 is involved in regulating the acetylation of mitochondrial proteins involved in ATP generation, and like Sirt1, appears to be increased in activity and number with SGLT2 inhibition.42,46
In cardiomyocytes, HFpEF pathology includes complex energy metabolic changes, with decreases in both glucose oxidation and mitochondrial oxidative capacity, resulting in decreased ATP production with cardiac remodelling, and this may be rescued by Sirt3 activity.84,85 Angiotensin II-induced HFpEF mice resulted in decreased glucose oxidation with reduced pyruvate dehydrogenase (PDH) activity, and Sirt3 has the ability to deacetylate and activate PDH.84,85 Related to this, increased microRNA-195 in failing hearts appears to inhibit Sirt3 by binding to its mRNA 3’-UTR, increasing the acetylation and deactivation of mitochondrial proteins, including PDH and ATP synthase.85 Therefore, increasing Sirt3 activity with SGLT2is may promote the deacetylation and activation of these enzymes, and improve mitochondrial respiration.42,46,85 This also suggests miR-195 as a potential target in HFpEF therapy through its relationship with Sirt3.44,85
In endothelial cells, Sirt3 may regulate a metabolic switch between glycolysis and mitochondrial respiration, protecting against coronary microvascular rarefaction – an emerging major contributor in HFpEF that involves endothelial cell apoptosis, cardiomyocyte hypoxia, titin-based cardiomyocyte stiffness and myocardial fibrosis.44,83,86 Endothelial cells rely predominantly on glycolysis for energy production, and Sirt3 deficiency in endothelial cells exhibit reduced glycolysis and higher levels of endothelial mitochondrial respiration, with increased production of ROS and increased acetylation of p53.83 Sirt3, which is increased with SGLT2 inhibition, may restore glycolytic predominance in endothelial cells, and also has potential to deacetylate p53, thus protecting against coronary microvascular rarefaction in HFpEF.83,87
In skeletal muscle, HFpEF is associated with a rapid depletion of high-energy phosphates early in exercise, contributing to exercise intolerance and early fatigue, hallmarks of HFpEF.88 Enhanced acetylation due to diminished Sirt3 may have inhibitory impacts on fatty acid β-oxidation enzymes in skeletal muscle, impairing mitochondrial oxidative capacity and contributing to exercise intolerance.89 Empagliflozin restored lowered exercise endurance capacity by activating skeletal muscle fatty acid oxidation in a murine model of HF.90 Importantly, dapagliflozin and canagliflozin improves symptoms and physical limitations in HFpEF, and beneficial skeletal muscle alterations may be involved with SGLT2 inhibition.11,14
Sirtuin 6
Sirt6 is predominantly a nuclear deacetylase that is downregulated in HF, and has demonstrated a role in protection against ventricular hypertrophy by modulating insulin-like growth factor (IGF)/protein kinase B (Akt) signalling.91 Sustained activation of IGF/Akt signalling due to hyperinsulinaemia, especially in the cardiometabolic phenotype of HFpEF, may lead to pathological hypertrophy through surplus energy signalling in cardiomyocytes.24,91 Sirt6 controls IGF/Akt signalling at the level of chromatin through the stress response transcription factor, c-Jun, and by deacetylation of H3K9.91 Thus, increased activity of Sirt6 may dampen IGF/Akt signalling and protect against this pathological hypertrophy.91 SGLT2is may also indirectly decrease signalling through this pathway by lowering serum insulin levels.24
SGLT2is Decrease Epicardial Adipose Volume and Actions of Pro-inflammatory Adipocytokines
In addition to the beneficial activation of sirtuins, the glucose-deprived state generated by blocking SGLT2 in the proximal tubule leads to increased serum glucagon and decreased serum insulin levels, thereby enhancing lipolysis with subsequent reduction of epicardial adipose tissue (EAT).36 Excess EAT especially contributes to the cardiometabolic phenotype of HFpEF as a mechanical obstacle to LV filling (pericardial restraint), and thereby aggravating diastolic dysfunction by enhanced diastolic ventricular interaction.36,44 The EAT also shares a common blood supply with the myocardium, with no fascia separating the two tissues, possibly allowing EAT-secreted pro-inflammatory (interleukin-6, interleukin-1β, tumour necrosis factor-α), pro-oxidant (H2O2, superoxide) and profibrotic (TGF-β) mediators to act on the surrounding myocardium – inducing extracellular matrix remodelling, hypertrophy and defective autophagic flux.44,92,93
Lipolysis of EAT may positively modify energy metabolism in the myocardium by providing it with free fatty acids and ketone bodies.36 Decreasing EAT can relieve pericardial restraint and improve end-diastolic pressure volume relationship in HFpEF, but also has other potential protective mechanisms due to the reduction of EAT-secreted adipokines. For example, decreased TGF-β and tumour necrosis factor-α may result in less diffuse interstitial fibrosis and hypertrophy with decreased LV stiffness – potentially contributing to the reduced LV mass on cardiac magnetic resonance demonstrated with SGLT2 inhibition, albeit this would be a longer-term mechanism versus the immediate effects of decreases in myocardial oedema due to osmotic diuresis (discussed earlier).36,72,94,95 Reduced TGF-β may also protect the atrial myocardium from fibrosis and AF.96 In addition, reducing EAT decreases superoxide production and its scavenging effects attributed to impairing the nitric oxide/cyclic guanosine monophosphate/protein kinase G pathway, resulting in decreased titin hypophosphorylation and a subsequent reduction in LV stiffness – potentially providing haemodynamic relief in HFpEF patients with a rightward shift in the end-diastolic pressure volume relationship.36,93,97,98
Notably, the EAT also secretes leptin, and circulating levels of leptin are increased in HFpEF.99,100 EAT-derived leptin induces myocardial remodelling in high-fat diet-induced obese rats, partly by stimulating upregulation of type I collagen via the JAK2/STAT3-TGF-β1/Smad3 pathway in cardiac fibroblasts.101 SGLT2i-induced increases of Sirt1, which has the ability to inhibit TGF-β1 signalling via deacetylation of Smad3 (discussed earlier), may mechanistically contribute to the ability of SGLT2is to counterbalance the deleterious actions of leptin-mediated fibrosis in HFpEF patients, especially in those who are obese. 102,103 Leptin may also increase oxidative stress in the heart through increased activity of NHE1, and SGLT2is have been shown to inhibit NHE1 (discussed later), thereby potentially protecting the heart against the paracrine effects of leptin-induced oxidative damage.104 Finally, it has been hypothesised in obesity-related HFpEF patients, that leptin enhances SGLT2 expression in the renal tubules and augments the secretion of aldosterone by the adrenal glands – leading to sodium retention and plasma volume expansion.103,105,106 The beneficial natriuretic actions of SGLT2is via inhibition of SGLT2/NHE3 and decreases in aldosterone levels are the opposite of the pathological sodium retention encouraged by leptin in HFpEF patients.103 Interestingly, decreased leptin levels seen with SGLT2 inhibition may not be only due to a reduction in adipose tissue, but could also be because of direct interference with leptin synthesis.107 This may explain why SGLT2i-induced decreases in leptin levels are out of proportion to their modest effects on weight loss.108
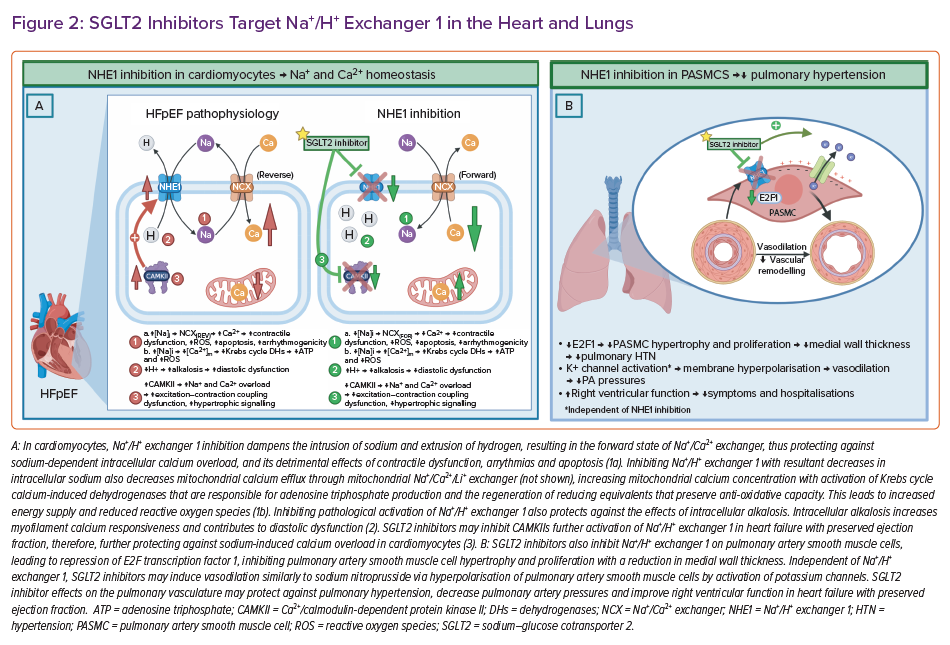
SGLT2s Target Na+/H+ Exchanger 1 in the Heart and Lungs
Increasing evidence supports SGLT2is’ inhibition of NHE1 in the heart, either directly and/or indirectly, explaining some of the cardioprotective mechanisms in HFpEF (Figure 2).38,109,110 Enhanced activity of NHE1 has been detected in HFpEF pathology, such as in pressure overload and hypertensive hypertrophy.111–113 When pathologically activated in cardiomyocytes, NHE1 exchanges hydrogen for an intrusion of sodium with reversal of the Na+/Ca2+ exchanger, resulting in sodium-dependent calcium overload, pH alterations and oxidative stress.59
Sodium-dependent calcium overload is injurious to cardiomyocytes, and leads to contractile dysfunction, arrhythmogenicity and apoptosis.114 Intracellular sodium overload also facilitates mitochondrial calcium efflux through the mitochondrial Na+/Ca2+ exchanger, impairing calcium-induced Krebs cycle dehydrogenases that are responsible for ATP production and the regeneration of reducing equivalents that preserve anti-oxidative capacity.115 This leads to inadequate energy supplies and to the deleterious release of ROS.115
Pathological activation of NHE1 also induces intracellular alkalosis, possibly increasing myofilament calcium responsiveness and contributing to diastolic dysfunction.116,117 Furthermore, NHE1 hyperactivation is associated with the activation of Ca2+/calmodulin-dependent protein kinase II, which phosphorylates and activates various sodium channels, including NHE1 – leading to further sodium and calcium overload, and oxidative damage.38 Calmodulin-dependent protein kinase II activity may play a role in the development and progression of HF by interfering with the fine-tuning of myocardial excitation-contraction coupling, and also by altering gene expression in hypertrophic signalling.118,119 Interestingly, mechanistic evidence supports SGLT2is’ inhibition of calmodulin-dependent protein kinase II, which could be responsible for the indirect inhibition of NHE1 by decreasing its phosphorylation.38,109
Further evidence for SGLT2is providing HFpEF patients benefit through NHE1 inhibition in the heart is shown by dapagliflozin ameliorating diastolic dysfunction, possibly by targeting coronary endothelium and cardiomyocytes through NHE1 in a non-diabetic model of HFpEF.120 Also, chronic inhibition of NEH1 attenuates cardiac hypertrophy, and prevents cellular remodelling after pressure and volume overloading in rabbits.114 A mechanistic study using artificial intelligence also showed empagliflozin-mediated NHE1 inhibition appears to modulate cardiomyocyte stiffness, myocardial extracellular matrix remodelling, systemic inflammation and concentric hypertrophy mechanisms.110 Validation of the data was performed by measuring declining plasma concentrations of inducible nitric oxide synthase, NLR family pyrin domain containing 3 inflammasome and TGF-β1 during 12 months of empagliflozin treatment.110
Finally, NHE1 inhibition in pulmonary artery smooth muscle cells may protect against pulmonary hypertension through repression of nuclear transcription factor, E2F transcription factor 1, leading to reduced proliferation and hypertrophy of pulmonary artery smooth muscle cells, and decreased medial wall thickness (Figure 2) in the pulmonary vasculature, as shown in mice.121 Interestingly, also in mice, SGLT2is have been shown to relax pulmonary vasculature in a dose-dependent manner akin to sodium nitroprussides (nitric oxide donor) mechanism – activation of potassium channels and hyperpolarisation of pulmonary artery smooth muscle cells.122 Clinically, empagliflozin has demonstrated rapid reductions in pulmonary artery pressures that were amplified over time.123 These SGLT2i-induced pulmonary vascular effects may aid in decongestion and improve right ventricular function in HFpEF patients, potentially contributing to the reduced symptoms and hospitalisations demonstrated in RCTs.9,41
Conclusion
SGLT2is may be the first drug class to improve cardiovascular outcomes in those suffering from HFpEF, providing hope to this heterogenous and difficult to treat population. RCTs are demonstrating a significant reduction in HF hospitalisations, with an improvement in functional status. Future trials are still needed to validate the effect of SGLT2is on mortality in HFpEF patients, and to determine their efficacy in patients at the higher end of the EF spectrum.
SGLT2is may provide a class effect by targeting a wide array of pathophysiological HFpEF pathways and mechanisms. Through inhibition of SGLT2 and NHE3 in the kidney, SGLT2is promote glycosuria, osmotic diuresis and natriuresis. The glucose deprivation is associated with a decrease in epicardial adipose volume, and SGLT2is reduce and oppose the actions of its detrimental adipocytokines. The glucose-deprived state also activates sirtuins – proteins that may protect the heart against oxidative damage, and positively regulate metabolism in the heart and skeletal muscle. Additionally, direct actions of SGLT2is via inhibition of NHE1 in the heart and lungs may protect against sodium-induced calcium overload and pulmonary hypertension, respectively.