







Heart failure (HF) is a feared endpoint for most cardiovascular diseases and is a major cause of morbidity and mortality. The worldwide prevalence of HF is between 2 and 3 % and rises sharply at around 75 years of age, so that the prevalence in 70- to 80-year-old people is between 10–20 %. With a 50 % 5-year survival rate, HF is predicted to be the leading cause of all morbidity by 2020.1,2 Despite the varied etiologies, ventricular dysfunction is ultimately the result of pathologic cardiac remodelling. Cardiac remodelling is defined as a series of compensatory alterations in the size, shape, and function of the myocardium in response to cardiac injury with the aim being to restore cardiac output. However, if this process continues, chronic cardiac stress magnifies maladaptive mechanisms, including cardiac hypertrophy, fibrosis, ventricular dilatation, alteration in geometry, chronic inflammation and increased cellular apoptosis, leading to a vicious cycle of deterioration of cardiac function and worsening of HF.3
The remodelling process involves the permanent cell types of the myocardium, namely the myocytes, the fibroblasts, the endothelial cells, the smooth muscle cells and the stem cells, but also transient cell populations such as immune and circulating stem cells.4 While the cardiac myocyte (CM) has been the focus of most HF research to date, increasing evidence has implicated the cardiac fibroblast (CF) as a key pathologic determinant in cardiac remodelling in both ventricles and atria.5 Dynamic interactions among the different cardiac cell populations via mechanical, chemical and electrical means, as well as their interactions with the extracellular matrix (ECM) determine cardiac physiology and pathology.4,5 Better understanding of these cell-to-cell and cell-to-ECM communications may provide potential novel therapeutic targets for the treatment of HF. In this review, the authors aim to explore the contribution of cellular cross-talk in the cardiac remodelling process.
Cellular organisation in the heart
Cardiac myocytes
The human heart contains an estimated 2–3 billion CM cells, which constitute about 75 % of the total volume of the myocardium, although only about one third of the total cell number.6,7 The major function of the CM is to carry out the cardiac contraction-relaxation cycle. Electrically, CM depolarise in response to signals from the sinoatrial node. Calcium is responsible for translation of the signal into muscular contraction, with calsequestrin in the sarcoplasmic reticulum being the major calcium-binding and storage protein. Mutation of this receptor can lead to a pathologic state of the myocardium, in which delayed after-depolarisation becomes prevalent.4,8
CM can act via chemical signalling by secreting various growth factors and cytokines.9,10,11,12 Moreover, they have been shown to exhibit a mechano-electrical feedback, in which mechanical force influences the electrical potential of the myocyte membrane.13
Cardiac fibroblasts
The majority of non-CM cells are CF. These are traditionally responsible for the maintenance of the structural integrity of the heart through regulation and turnover of the ECM. Strictly-controlled production and secretion of proteins, such as collagens, fibronectin, matrix metalloproteinases (MMPs), and tissue inhibitor of metalloproteinases (TIMPs), form a highly organised three-dimensional network surrounding myocytes and allow for mechanical force distribution throughout the myocardium.14 CF are cells of mesenchymal origin, but arise also from the fibrocytes, bone marrow-derived cells in the neonatal and adult heart.15,16,17 The main features of CF are the lack of a basement membrane, distinguishing them from all other permanent cardiac cells; an extensive Golgi apparatus; a relatively large endoplasmic reticulum, which underpins their role in protein synthesis and secretion; and their flat, spindle-shaped morphology with multiple filopodia originating from the main cell body.4
Under pathological stress, the resting fibroblast transdifferentiates into myofibroblast, expressing characteristics of smooth muscle cells and an enhanced capability for migration and tissue invasion.18,19,20 Moreover, myofibroblasts have been shown to originate from alternative cellular sources, including the local mesenchymal tissue, smooth muscle cells, vascular pericytes, a myeloid lineage, and fibrocytes.21,22 To date, the precise contribution of each cellular source to the myofibroblast population is poorly understood, but one hypothesis is that the transformation of differential cellular sources into myofibroblasts accounts for the manifestation of different forms of fibrosis (chronic versus acute).5 Several humoral factors can affect fibroblast activation and secretion of ECM proteins as well as their differentiation into myofibroblasts, including angiotensin II (AngII), endothelin 1 (ET-1), transforming growth factor- (TGF- ), fibroblast growth factor 2 (FGF2), and insulin-like growth factor-1 (IGF-1).17,23,24,25 Myofibroblasts play an important role in reparative healing of the myocardium following tissue damage.26 However, imbalanced myofibroblast activity results in interstitial fibrosis, ventricular stiffening, remodelling and failure.27 An important therapeutic consideration is the degree to which cardiac fibrosis is reversible and whether myofibroblasts can undergo senescence and apoptosis or dedifferentiate back to their original cell type.5 There is evidence that two weeks after myocardial infarction about a third of myofibroblasts undergo apoptosis; nevertheless, the fate of the remaining cells remains in question.28 The current research for fibrosis is directed at understanding and inhibiting signalling pathways that regulate myofibroblast transformation.
As with CM, fibroblasts also participate in mechano-electrical signalling which accounts for changes in the contractile function of the heart and arrhythmiogenicity in response to cardiac load alterations.13
Endothelium
The endothelial cells (EC) form the inner lining of blood vessels and as recently as the first half of the 20th century were viewed simply as barriers of blood flow. Today, endothelium has been recognised as a dynamic organ with complex biological functions, including the control of vascular permeability, the vasomotor control of coronary arteries, the regulation of haemostasis, immune responses and angiogenesis. The EC release nitric oxide, ET-1, AngII, prostaglandins, pro- and anticoagulant factors and growth factors that can affect the myocardial and vascular function.29 Furthermore, endothelium plays an important role in the regulation of heart size.30 There is evidence that the increase in myocardial vasculature is not only supportive of CM hypertrophy, but may actually induce the relevant process.31 Several studies support the notion that an increase in the capillary density is important for the development of physiological cardiac hypertrophy, whereas a reduction of the vascular bed size contributes to HF decompensation.32,33,34
The Extracellular Matrix
The cardiac ECM is a complex architectural network consisting of a variety of proteins, including fibronectin, fibrillin, periostin, more than 28 different types of collagen, glycoproteins (e.g. thrombospondins, secreted protein acidic and rich in cysteine, tenascins), proteoglycans (e.g. versican, syndecans, biglycan), and glycosaminoglycans (e.g. hyaluronan, heparan sulphate), creating strength and plasticity.35,36 Fibronectin is a multi-domain protein that interacts with proteoglycans and collagen to mediate cellular function. Periostin interacts with other components of the ECM, as well as with fibroblasts, playing a role in their differentiation. Collagen is crucial for maintaining the elasticity and integrity of the heart. Glycoproteins, proteoglycans and glycosaminoglycans are upregulated on cardiac injury and control key processes in the remodelling of myocardium, such as inflammation, fibrosis, and angiogenesis. The ECM is able to store and release a number of growth factors, chemokines and cytokines.4,35 When tissue is injured, there is an increased expression of MMPs and increased degradation of the ECM to promote healing and scar formation. Excessive degradation is blocked by endogenous TIMPs. In fact, there is a delicate balance between MMPs and TIMPs that contributes to the regulation of the cardiac remodelling process37.
Cardiac Cell–cell and Cell–ECM Communications
Cardiomyocyte to Cardiomyocyte Communication
There are many routes of CM–CM communication, including the secretion of autocrine factors and direct contact via gap junctions and adhesion complexes. Autocrine cross-talk is carried out by myocyte- secreted factors that include leptin, hepatocyte growth factor, endothelin-1 and FGF and TGF- family members.29 Gap junctions allow CM-CM communication via the exchange of ions and small solutes. In the myocardium, gap junction proteins of the connexin family have been shown to play a crucial role in determining impulse conduction and the heart morphogenesis.38 Adhesion-complex communications include intracellular signalling cascades that are triggered by cell–cell or cell–ECM engagement of specific proteins in these complexes. This type of communication may alter CM responses to growth factors leading to myocardial hypertrophy.29
Another factor released by the CM upon acute myocardial infarction is tumour necrosis factor-alpha (TNF-alpha).39 TNF-alpha has been found to be mainly released via hypoxia-inducible factor 1-alpha- mediated non-classical secretory pathways, involving release of vesicles containing a membrane variant of TNF. The identified vesicles were suggested to be exosomes and, when obtained from hypoxic CM, could trigger cell death in other CM.40,41
Cardiac Fibroblast to Myocyte Communication
CM and CF are spatially intermingled in the myocardium with virtually every CM bordering one or more CF. Bidirectional communication between CF and CM can be mediated by paracrine signals, direct cell- cell interactions and indirect interaction via ECM.42
Both CF and CM secrete many different chemokines, cytokines, growth factors and other soluble agents that play a key role in cardiac physiology and pathophysiology.42 Connective tissue growth factor (CTGF), which is induced by TGF- and expressed in both CM and CF, has been associated specifically with CF proliferation and ECM production in the setting of myocardial fibrosis.43,44 CTGF expression is negatively regulated by two cardiac microRNAs (miRNAs), miR-133 and miR-30.45 miRNAs are non-coding RNAs that regulate messenger RNA (mRNA) translation or degradation. They can be actively secreted or passively leaked from a cell to act either on an adjacent cell or on distant cells along the vasculature.46 While miR-30 is expressed in both CF and CM, miR-133 is expressed specifically in CM. miR-133-knockout mice develop excessive fibrosis and HF while miR-133 knockdown also causes cardiac hypertrophy with impaired cardiac function.47,48 CF also secrete interleukin 33, which cross-regulates CM in vitro, reduces pressure-overload hypertrophy and fibrosis, and improves cardiac function and survival after myocardial infarction in vivo.49,50 Interleukin 6 (IL-6) induces cell proliferation, protects cells from apoptosis, promotes ECM turnover, and causes cardiac hypertrophy.51 It is of note that interactions between CM and CF markedly promote the secretion of IL-6, as shown by increased levels in conditioned medium from CM-CF co-cultures compared with CM or CF only cultures, indicating that communication between the two cell types is required for the secretion of some paracrine factors.52,53 Platelet-derived growth factor (PDGF) has been shown to play an important role in cardiac fibrosis and angiogenesis via its binding to protein tyrosine kinase receptors. The latter control fibroblast proliferation and migration and are also linked to ECM deposition.4 In transgenic mice, PDGF expression can be largely enhanced, leading to dilated cardiomyopathy and HF.54 IGF-1 secretion by fibroblasts also plays a key role in mediating the adaptive response of myocardium to pressure overload.55 Moreover, paracrine signals released from CF may affect the expression and function of ion channels and gap junctions in CM.56,57,58,59 Apart from the conventional exocytosis of paracrine factors, there is evidence that a pannexin- based mechanism exists in the myocardium. Pannexins were found to form functional channels in single membranes that can make possible paracrine intercellular communication by releasing ATP and other small molecules from the cytoplasm to the extracellular space.42
CM and CF are able to communicate electrically through connexin- mediated gap junctional connections.4 CF are not electrically excitable, but their membrane contains ion channels.60,61,62 Without coupling to CM, CF operate as passive electrical insulators.63 However, when coupled to CM, they can affect action-potential characteristics and conduction velocity in CM.42,64 CM–CF coupling has also been shown to alter intercellular calcium cycling alternans, which could play an additional role in arrhythmogenesis in fibrotic heart tissue.65 Another mechanism for mechanical coupling between CM and CF is through adherens junctions and the cadherin–catenin complex at their core.42 Cadherins are transmembrane receptors that bind adjacent cells, link intracellularly to actin and intermediate filaments via catenins and facilitate bidirectional transmission of cytoskeletal tension between cells.66,67 Cadherin staining has been detected between co-cultured CM and myofibroblasts.68,69 It has been shown that TGF –activated myofibroblasts can exert tonic contractile forces on CM and slow electric propagation as a result of increased mechanosensitive channel activation.69 A more recently described route of cell–cell cross talk that permits intercellular communication over longer distances is via membrane nanotubes. It has been demonstrated that organelles and cytoplasmic proteins exchange and calcium signal propagation occurred from CM to CF and vice versa through long, thin membrane nanotubular structures containing actin and microtubules.70,71,72
Cardiac Fibroblast to Endothelial Cell Communication
A number of studies have demonstrated that CF can interact with EC and modify the expression of both pro- and anti-angiogenic factors. CF-secreted growth factors, including vascular endothelial growth factor (VEGF) and FGF, act on EC and stimulate angiogenesis.4
However, FC can also act in an inhibitory way. It has been shown in vitro that pigment epithelium-derived growth factor can be expressed by CF and can inhibit VEGF-induced tube formation.73 Moreover, the CF-secreted MMPs and TIMPs are involved in the process of angiogenesis and under certain conditions they can lead to promotion or inhibition of tube formation.74,75,76
CF and EC can also communicate by exchanging miRNAs. There is evidence that multiple miRNAs are expressed in endothelial cells and can lead to the promotion or inhibition of angiogenesis.77,78
Cardiac Myocyte to Endothelial Cell Communication
In the heart, EC outnumber CM at nearly a 3:1 ratio with virtually every CM bordering one or more capillary.79 CM paracrine signalling plays a key role in the dynamic regulation of the vascular tone but may also affect long term growth and development of coronary arterial, venous and lymphatic trees.29 Among the multiple paracrine signals the most important are VEGFs. When there is either an overexpression or a deficit in VEGF, the ultimate result is cardiac dysfunction.80 It is of note that although CM represent less than a third of the total cell number in the heart, cardiomyocyte-specific deletion of the VEGF-A gene results in a decrease in the entire VEGF mRNA synthesis to less than 15 % of normal, emphasising the role of CM as the main source of this growth factor in the myocardium.29 When mice were engineered with cardiac myocyte-specific deletion of VEGF, the result was thinned ventricular walls, decreased contractile function and lack of neural stimulation.81 VEGF is considerably increased in the ischemic myocardium, whereas the vasculature in ischemic myocardium is more sensitive to VEGF- induced vasodilatation.82,83
CM–EC communication is very important for the vascular adaptations that occur during cardiac hypertrophy. Myocardial hypertrophy induced by expression of Akt1 is a clinical setting that illustrates the importance of the balance between cardiac and vascular growth. Transgenic overexpression of activated Akt1 in CM resulted in a varied spectrum of phenotypes from myocardial hypertrophy with preserved systolic function to ventricular dilatation and failure.84 In a tetracycline-inducible cardiac myocyte-specific Akt1 transgenic mouse model, short-term (two weeks) induction of Akt1 expression resulted in physiological hypertrophy that was accompanied by analogous myocardial angiogenesis.85 However, Akt1 activation for longer periods of time resulted in a disproportionate increase in cardiac mass compared with the extent of angiogenesis and development of HF, presumably on the basis of an inadequate blood supply to cover the requirements of the hypertrophic myocardium.85 CM and EC are able to communicate electromechanically through gap junction proteins, such as Cx43.86 and it has been demonstrated that VEGF can affect Cx43 expression in CM, suggesting another VEGF-dependent pathway as a mode of interaction between myocardium and vasculature.80
Cell to Matrix Communication
The principal mediators of molecular dialogue between a cell and its extracellular matrix environment are integrins.87 They are heterodimeric cell-surface molecules that mediate signalling from the extracellular space into the cell through integrin-associated signalling and adaptor molecules such as focal adhesion kinase, integrin-linked kinase particularly interesting new cysteine-histidine rich protein and non-catalytic (region of) tyrosine kinase adaptor protein-2.87 Via these molecules, integrin signalling interacts with receptor tyrosine kinases signalling to regulate survival, proliferation and cell shape as well as polarity, adhesion, migration and differentiation. In the myocardium and blood vessels, the function and regulation of these molecules can be partially disturbed, leading to cardiovascular diseases such as cardiac hypertrophy and atherosclerosis. Integrin-mediated cell adhesion can be modulated by membrane-associated proteins such as the ADAMs (a desintegrin and metalloprotease). ADAMs can alter expression and function of growth factor receptors and, as a result, will then affect several biological processes, including angiogenesis and cardiac hypertrophy.29,88
Conclusions and Therapeutic Perspectives
Each cell type in the myocardium interacts with another in various ways and has its own unique changes to gene and protein expression, protein secretion and response to signals from other cells (Figure 1). It has become clear that the myofibroblast is a significantly more complex cell type that was initially appreciated. Moreover, the ECM has emerged as a dynamic component in both the normal and diseased heart. Despite recent progress, our understanding of many of these cell–cell and cell–ECM interactions remains unclear.
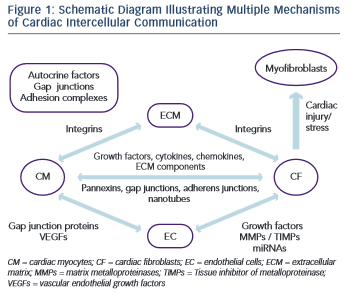
Cytokine antagonists, particularly those targeting TNF , did not provide any apparent clinical benefit in HF patients, despite the promising preclinical data, but targeting the role of inflammatory cytokine signalling in HF has provided fruitful lessons for future drug development. Moreover, it would be highly beneficial to identify pharmacologic agents that might inhibit myofibroblast formation in chronic disease states while sparing their protective role in physiological wound healing. One particularly important aspect of cell–cell cross-talk in the heart is the CM–EC interactions. Once our understanding of how changes in endothelial cell mass affect cardiac hypertrophy improves, we may gain more insight into the possibility of stimulating angiogenesis and vessel growth in the injured myocardium.
Further research into the dynamic intercellular communication is crucial for the development of novel therapeutic strategies that target directly HF disease progression.