










Fabry’s disease (FD) is a multisystem X-linked disease caused by pathogenic variants in the galactosidase-α (GLA) gene, leading to reduced α-galactosidase A (α-Gal A) enzyme activity.1 This reduction is responsible for the progressive lysosomal storage of globotriaosylceramide (Gb3) and related globotriaosylsphingosine (lyso-Gb3) in several tissues and organs.1
The clinical manifestations of FD depend on the residual α-Gal A enzyme activity, and two different subtypes of FD have been described, named the early-onset and later-onset phenotypes.1,2 Affected patients with the early-onset phenotype have low or no functional α-Gal A enzyme activity, with marked Gb3 and lysoGb3 accumulation and symptoms onset in childhood or adolescence, while those with the later-onset phenotype have residual α-Gal A enzyme activity, which is responsible for a milder phenotype and a later clinical presentation (rarely before the third decade).1
Cardiac involvement is the main determinant of adverse outcomes in patients with FD.3 It is largely variable in patients and ranges from uncomplicated asymptomatic disease to severe left ventricular hypertrophy (LVH), leading to disease complications, including heart failure and life-threatening arrhythmias, and premature death.3,4 The heterogeneous and often subtle presentation of FD can be responsible for a significant diagnostic delay, requiring active screening of high-risk patients.5,6
It is common for patients with the later-onset disease to present with single-organ involvement, mainly of the heart (usually manifesting as LVH or arrhythmias), central nervous system (manifesting as cryptogenic ischaemic stroke) or kidneys (manifesting as proteinuria or progressive chronic kidney dysfunction).1 Therefore, identifying specific clinical, laboratory or imaging findings in these patients may prompt a diagnostic work-up for FD.6–8 The main diagnostic steps are the identification of reduced α-Gal A enzyme activity, which is usually measured in plasma, leukocytes or dried blood spots, and confirmation of the disease-causing mutation in GLA.1 However, given that α-Gal A enzyme activity could be in the normal range in female patients, identification of a GLA mutation is required for the diagnosis.9
Different pathophysiologically driven therapies are currently or will soon be available to treat FD, with the most significant benefit observed in the early stages of the disease.10 Thus, the early diagnosis and risk stratification for the adverse outcome is crucial to determine when to start an aetiological treatment.11
This review describes the cardiovascular involvement in FD, focusing on the advances in diagnostic strategies, outcome prediction and disease management.
When to Suspect Fabry’s Disease
The high variability in the clinical manifestation of FD, with the different possible ages of onset and symptoms onset, can lead to delayed diagnosis and treatment.5 Therefore, given that FD is a multisystem disease, cardiologists, neurologists, dermatologists, nephrologists, paediatricians and ophthalmologists should all be aware of the possibility of FD, depending on the patient’s clinical presentation.6
In the cardiology environment, a diagnosis of FD should be systematically considered in the case of unexplained LVH, especially when it is concentric, symmetric, homogeneous and non-obstructive (Figure 1).3 All patients with LVH should be screened for additional extracardiac or cardiac manifestations associated with FD.7,8,12,13
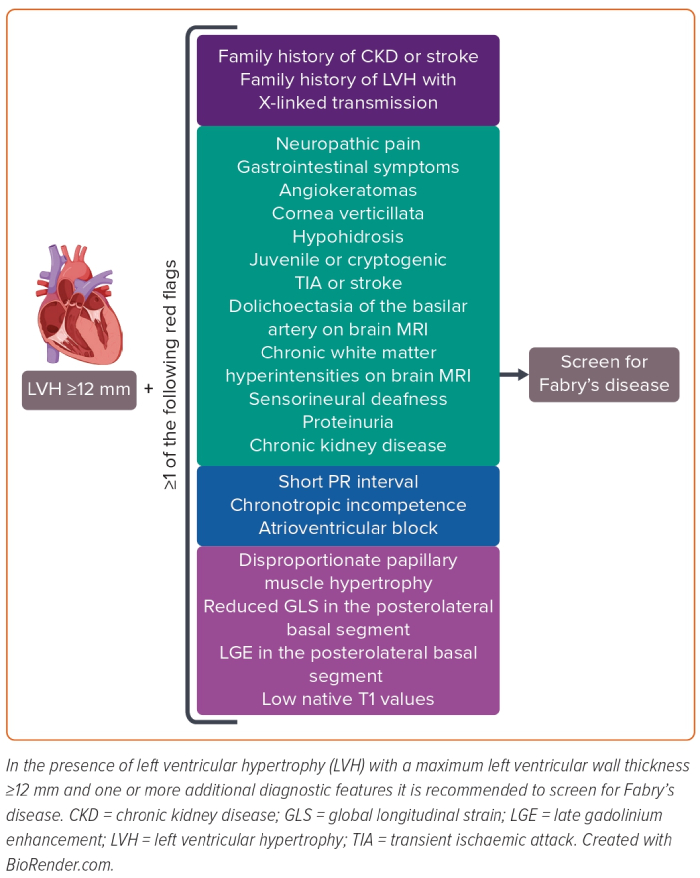
Extracardiac Red Flags
The clinical manifestations generally vary according to the age of presentation.
In patients with early-onset disease, the first symptoms include chronic neuropathic pain and episodic pain crises that generally emerge during childhood.14 Other common symptoms manifesting during childhood are gastrointestinal symptoms, including abdominal pain, diarrhoea, nausea or vomiting.15 However, these symptoms are common in the general population and are not specific to FD. Angiokeratoma is the most common dermatological abnormality and is a specific clinical feature of early-onset FD.16 Finally, hypohidrosis and corneal opacity (i.e. cornea verticillata) are other common manifestations during the first two decades of life.1
Cardiac, kidney and central nervous system involvement generally appear from the third decade and may be preceded by the other described clinical manifestations (in early-onset disease) or present as an isolated feature (in later-onset disease). For example, kidney involvement presents with progressive proteinuria and reduction in glomerular filtration rate, while cerebrovascular manifestations include transient ischaemic attack, stroke or isolated neuroradiological findings such as chronic white matter hyperintensities or basilar artery dolichoectasia.17–19 These features usually occur at an early age compared with the general population.
Cardiac Red Flags
In patients with LVH, several ECG, echocardiographic and cardiac MRI (CMR) findings may suggest FD.3,4 Typical ECG findings are short PR interval, atrioventricular block and diffuse repolarisation abnormalities.20 Common imaging findings are papillary muscle hypertrophy, right ventricular hypertrophy, advanced diastolic dysfunction, late gadolinium enhancement (LGE) in the basal inferolateral wall, and mitral and aortic regurgitation due to leaflet involvement.21
In addition, it is common to observe the so-called ‘binary sign’, characterised by a bright endocardial layer and a hypoechogenic intraventricular septum. The binary sign, previously considered specific for FD, has been observed in several other causes of LVH and is now considered to have low sensitivity and specificity for FD.22
All of the above-mentioned cardiac features have generally appeared in patients with advanced cardiac involvement, in whom the initiation of an aetiological treatment was not associated with significant benefit. Therefore, efforts have recently been directed to improve the identification of patients with early cardiac involvement.
Detection of Preclinical Cardiac Involvement
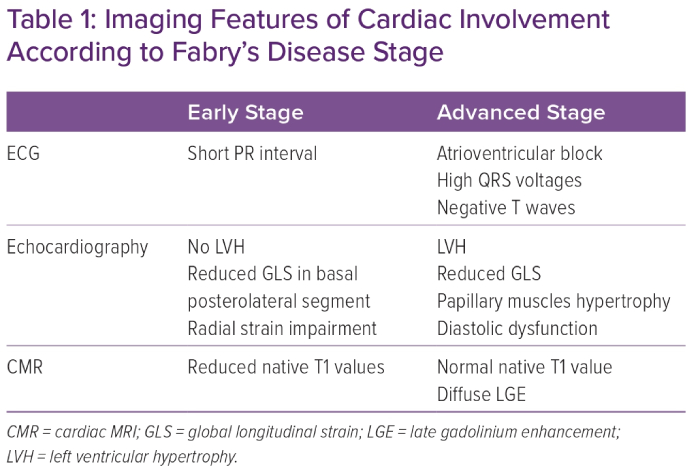
Given that the most significant benefit of aetiological treatment has been observed in patients with the mild disease phenotype, it is crucial to identify patients during the preclinical phase of the disease. Several ECG and imaging markers could be identified prior to LVH development (Table 1).
ECG Parameters
Several ECG abnormalities have been described in patients with FD and they generally correlate with the myocardial involvement phenotype.3 The typical ECG abnormality is the presence of a short PR interval, caused by the shortening of the P wave, and lacking the typical pre-excitation pattern.23 This sign is common in the early stages of the disease. In contrast, signs of LVH, negative T waves, and atrioventricular and intraventricular conduction delays generally appear in advanced stages.24
Thus, in the pre-hypertrophic phase, in which patients do not show signs of LVH or impaired left ventricular function, ECG abnormalities may represent the only signs of left ventricular involvement.25 Moreover, a significant correlation between ECG abnormalities and the detection of low native T1 values at CMR has been observed.26 Based on this observation, an ECG-based score to estimate the probability of detecting FD cardiac involvement at CMR has been proposed.26 This score was based on four different parameters: the Sokolow–Lyon index, the ratio between the P wave and PR segment duration, the QRS duration, and the QT duration, and can potentially improve the early detection of FD cardiac involvement.26
Echocardiographic Deformation-based Markers
Deformation-based parameters in strain analysis are superior to standard echocardiographic measurements in demonstrating left ventricular involvement.27 Left ventricular involvement in patients with FD shows a typical regional distribution from the early stages. Compared with the general population, FD patients have a significant reduction in average global longitudinal strain (GLS), and basal and mid-ventricular longitudinal strain.28 Moreover, patients with higher lysoGb3 values had lower apical–ventricular longitudinal strain values, suggesting an early basal and mid-ventricular left ventricular involvement. In contrast, the apical involvement was observed in the advanced stages of the disease.28
The left ventricular segments most affected in FD are the basal lateral and posterior segments. A correlation has been seen between the reduction of GLS and the amount of LGE at CMR.29 Indeed, the GLS reduction in the basal posterolateral segments was an independent predictor of LGE.29 Even in the absence of left ventricular involvement on standard echocardiography (i.e. normal left ventricular mass and preserved systolic function), GLS is reduced in FD patients compared with controls, especially in the basal segments. In addition, basal longitudinal strain reduction in FD was associated with major adverse cardiovascular events.30
The Gb3 and lysoGb3 accumulation process shows a typical myocardial mural involvement. Thus, in 33 newly diagnosed FD patients, layer-specific myocardial deformation analysis on 2D-speckle tracking (2D-ST) echocardiography showed a significant reduction in subepicardial compared with subendocardial longitudinal strain, with a consequent significant strain gradient. Furthermore, the subepicardial longitudinal strain was the best parameter for discriminating FD patients from healthy subjects.31
In addition, left ventricular radial and circumferential strain analysis has been investigated in recent years. Spinelli et al. showed that FD patients with preserved left ventricular ejection fraction (LVEF) had lower longitudinal, radial and circumferential strain than controls, regardless of left ventricular geometry.32 Moreover, in patients without LVH, the radial strain was significantly impaired, suggesting the possible role of radial strain as an early marker of left ventricular involvement in FD.32
Right ventricular (RV) involvement in FD on strain imaging has been studied more recently. In a recent study, RV systolic function assessed using non-deformation parameters was normal in approximately 92% of patients, while RV GLS and free wall strain (RV-FWS) were impaired in approximately 40%.33 Both strain-derived parameters were significantly more impaired in patients with LVH than in patients with normal cardiac mass, suggesting a possible association of RV involvement with the accumulation burden and the stage of the disease.33 Moreover, RV function assessed on strain analysis provides a better prognostic assessment than other RV echocardiographic parameters.
In a recent study of 56 patients with FD, RV-FWS was associated with adverse cardiovascular outcomes during a median follow-up of 47 months.34 However, RV-FWS did not retain a significant association with outcomes when adjusted for left ventricular GLS or left atrial volume index (LAVI), demonstrating the superior prognostic power of echocardiographic left-sided parameters in FD patients, and underscoring the fact that the prognosis is mainly driven by the severity of left ventricular cardiomyopathy.34
Cardiac MRI Features
CMR has a dominant role in the diagnosis, risk stratification and detection of preclinical myocardial involvement in patients with FD.11 Moreover, it offers the possibility to identify several diagnostic clues suggestive of FD, even in the pre-hypertrophic phase.35 With the use of gadolinium contrast agents, it is possible to observe in many patients with FD the presence of mid-myocardial LGE in the basal inferolateral wall. This typical pattern is helpful for the differential diagnosis of LVH.35 Moreover, there is increasing evidence that LGE may also be observed before LVH development, enabling early identification of patients who may benefit from aetiological therapies, especially with regard to female patients.36
In addition, CMR can provide myocardial tissue characterisation. Low native T1 values appear to be sensitive for identifying patients with FD, correlate with intracellular Gb3 accumulation, and represent an early marker of the disease in the pre-hypertrophic phase. Three stages of cardiac involvement have been proposed using three CMR features: native T1 value; LGE; and LVH.24 In the first stage (the accumulation phase), patients have low native T1 and do not present LGE or LVH. In this phase it is common to observe ECG abnormalities. In the second stage (the inflammation and myocyte hypertrophy phase), patients have low native T1 and LGE, with or without LVH. Finally, in the third stage (the fibrosis and impairment phase), patients have LVH, extensive LGE and normal T1.
Finally, CMR can potentially improve the risk stratification of patients with FD. It can be used to identify risk factors, such as diffuse LGE, extreme LVH and left ventricular dysfunction, which are associated with the increased risk of adverse events. Moreover, an internally validated model for predicting outcomes in patients with FD has been proposed.37 Three different clinical parameters, that is, age, left ventricular mass index and T1 dispersion, are incorporated into the model, showing the potential for it to be applied without the need for gadolinium contrast agents.37
Major Cardiovascular Disease Complications
The hypertrophic cardiomyopathy phenotype is the most common cardiovascular manifestation of FD.38 However, evolution to restrictive cardiomyopathy may occur in advanced stages of the disease.39 Therefore, FD should be considered in the differential diagnosis of patients presenting with heart failure (HF) with preserved ejection fraction (HFpEF).10 Moreover, cardiac involvement may eventually progress to left ventricular systolic dysfunction and HF with reduced ejection fraction (HFrEF) in 6–8% of patients (especially in the absence of enzyme replacement therapy) and confers a high risk of HF-related mortality. Patients with FD have been shown to have a significant risk of developing overt HF, which was observed in 23% of patients, usually between the third and the fifth decades of life.40 Furthermore, progression to advanced HF has been observed in 10% of patients over a median period of 7.1 years, and increased levels of cardiac biomarkers (i.e. troponin T, N-terminal pro-B-type natriuretic peptide) and a greater extent of fibrosis have been associated with a reduction in LVEF during the follow-up.40 However, published studies are largely heterogeneous regarding the definition of adverse outcomes. Therefore, prognostic models that accurately predict HF development or HF-related death are lacking.
Treatment
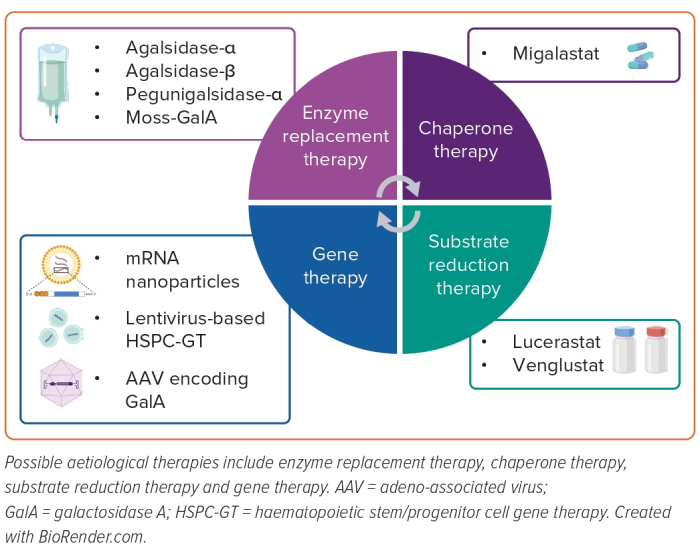
The current treatment options for FD include enzyme replacement therapy (ERT) and oral chaperone therapy with migalastat. However, several different therapeutic options will be available soon (Figure 2). The decision to start an aetiological treatment varies according to the sex and clinical manifestation of the disease.
According to the current expert recommendations, the treatment of FD should be considered in all male patients, regardless of the clinical manifestation.2,6,41 In contrast, female patients should receive an aetiological therapy after identifying any signs of organ involvement, while those without signs of organ involvement will require only serial follow-up. Thus, after a diagnosis of FD, a multidisciplinary team should be consulted for further evaluation and treatment tailoring.
Enzyme Replacement Therapy
ERT has been the cornerstone of FD management since the early 2000s.42,43 Two formulations are currently available: agalsidase-α 0.2 mg/kg bodyweight and agalsidase-β 1.0 mg/kg body weight. Both are given as biweekly infusions for lifelong therapy and are effective in clearing Gb3. Nearly every aspect of FD is addressed by ERT, with several studies reporting stabilisation of renal function, slowed progression of myocardial fibrosis and hypertrophy, reduction in thromboembolic episodes, and improvements in symptoms and quality of life.44–51 Early therapy is required to prevent or minimise disease development.47,52,53 Patients who begin ERT at a younger age have better results. Conversely, as the disease advances, ERT efficacy declines.
However, ERT has some drawbacks. Frequent infusion reactions, high cost, and the burden of a lifelong regimen are important issues that need to be addressed. Moreover, a significant proportion of patients, especially male patients lacking native α-Gal A, develop antidrug antibodies (ADAs). ADAs may work at different levels, limiting the effects of infused enzymes. These neutralising antibodies may bind different sites, preventing cellular uptake and the intracellular conformational changes required for enzymatic function.54
Despite these limitations, research on ERT has continued. Various attempts to refine the pharmacokinetic properties of infused enzymes have paved the way for next-generation ERT, with a focus on biodistribution. Infused enzymes are mostly taken up by the liver, while cardiomyocytes and podocytes have low intracellular levels.55
Pegunigalsidase-α (PegA) (previously PRX-102) is manufactured in modified tobacco cells.56 PEGylation is a chemical modification that offers attractive benefits. It hides molecules from the immune system, delays clearance and increases half-life.57 These features may help counteract ERT’s immunogenicity and lengthen the intervals between infusions.57 Preclinical studies have shown greater heart and kidney uptake than first-generation ERT.55 This potentially ground-breaking preclinical research was followed by three randomised clinical trials (RCTs).58–60 First, patients with worsening renal failure despite long-term ERT with agalsidase-β were enrolled in the BALANCE (NCT02795676) trial for the first head-to-head comparison between first- and second-generation ERT.58 Second, the feasibility of monthly 2 mg/kg PegA infusion was evaluated in the BRIGHT (NCT03180840) study.59 This regimen was well-tolerated in FD patients previously treated with standard ERT. Finally, the safety and effectiveness of switching from agalsidase-α to 1 mg/kg biweekly PegA infusions were tested in the BRIDGE (NCT03018730) study.60 After only 6 months from ERT transition, an improvement in renal function was seen.
Moss-GalA is another potential second-generation ERT. According to experimental models, it is taken up by mannose receptors, especially in the kidney. Moreover, it is stripped of immunogenic potential.61 The safety and pharmacokinetics were assessed in female patients not receiving ERT for at least 3 months.62 However, other studies are expected to corroborate these findings.
Chaperone Therapy
The only approved chaperone therapy is migalastat, an oral pharmacological chaperone that enhances native α-Gal A activity.63 This effect is provided by a specific and reversible interaction with the catalytic sites of amenable mutant forms of α-Gal A. More than 1,000 mutations have been identified.2 Some of them have minimal residual activity and are amenable to migalastat therapy.64 Once migalastat binds to α-Gal A, the enzymatic structure is stabilised, degradation in the endoplasmic reticulum is avoided, and appropriate lysosomal trafficking is promoted. Then, lower lysosomal pH levels lead to migalastat detachment, enabling α-Gal A to clear Gb3. Finally, migalastat is swiftly shuttled out of the cell and eliminated.2
The efficacy and safety of oral 123 mg migalastat, given every other day, has been evaluated in two pivotal Phase III RCTs: FACETS and ATTRACT.64,65 ERT-naive and ERT-treated patients, respectively, were enrolled. Besides the oral treatment route, other advantages include a lack of immunogenicity and the small size, which can potentially enable the crossing of the blood–brain barrier.
Substrate Reduction Therapy
Substrate reduction therapy (SRT) represents a paradigm shift in FD management. Unlike ERT and migalastat, SRT operates by preventing the accumulation of metabolites that cannot be broken down because of the underlying enzyme deficiency.66 SRT inhibits glucosylceramide synthase (GCs), the enzyme responsible for catalysing the first step in glycosphingolipid biosynthesis.66 Moreover, SRT may be given orally, regardless of genotype.
Gaucher’s disease was the first lysosomal storage disease to be treated with SRT.67 There are two approved SRTs for Gaucher’s disease: miglustat, a glucose-based iminosugar, and eliglustat, a ceramide analogue.68 These two therapies laid the groundwork for novel FD compounds, which are currently being investigated in RCTs.
Lucerastat, the galactose derivative of miglustat, significantly reduced Gb3 levels in mice and FD patient-derived fibroblasts.66,69 A phase III RCT, MODIFY (NCT03425539), was conducted to investigate the effects on neuropathic pain.70 Unfortunately, no differences in the primary endpoint were observed. However, most patients entered the open-label extension (NCT03737214), and a recent interim analysis showed further reductions in Gb3, and slowing of estimated glomerular filtration rate decline and cardiac hypertrophy, encouraging long-term evaluation.
Venglustat is a ceramide-derived iminosugar with the unique property of being able to cross the blood–brain barrier.71 A Phase IIa study (NCT02228460) assessed the safety profile and pharmacological features and explored efficacy outcomes in ERT-naive classic-FD patients. After 26 weeks, patients could choose to participate in an open-label extension (NCT02489344) of the study.72 Daily intake of 15 mg of venglustat reduced several biomarkers (such as Gb3, GC, GM3 ganglioside, and lyso-Gb3), suggesting that both synthetic and catabolic glycolipid pathways are affected. Moreover, it prevented the progression of FD with an acceptable rate of adverse events. Indeed, light microscopy evaluation of skin biopsies indicated significantly lower Gb3 scores after venglustat treatment. Additionally, a quantitative analysis of Gb3 inclusions in superficial skin capillary endothelium corroborated the light microscopy results. Two phase III RCTs, PERIDOT (NCT05206773) and CARAT (NCT05280548), are ongoing and are investigating the effects of venglustat on neuropathic pain and LVH, respectively.
Despite the established efficacy of approved medications and promising results of novel treatments, none is curative. Therefore, a substantial unmet medical need exists with regard to FD patients.
Gene Therapy
Many gene delivery strategies, including viral and non-viral approaches and in vivo and ex vivo methods, have been studied. This field of research is based on the idea that the treated cells would overexpress and produce α-Gal A, which will be picked up by other cells via the mannose-6-phosphate receptor.73 Endogenous enzymatic expression in specific tissues may be achieved using DNA and RNA-based delivery systems. Efficacy was demonstrated for mRNA nanoparticles. Interestingly, a dose-dependent trend was noted. Regrettably, owing to the short half-life of mRNA and its non-genomic action, repeated injection is needed.74,75
Haematopoietic stem/progenitor cell gene therapy (HSPC-GT) has the potential to permanently cure FD, avoiding or at least reducing the need for repeated treatment. Transduction of autologous haematopoietic stem and progenitor cells (HSPCs) can be achieved using a lentivirus with an active integrase enabling ex vivo insertion of a transgene. After insertion, engineered HSPCs are then infused and start to replicate. In this way, the inserted transgene is expressed by the progeny of the HSPCs. In addition, transgene products can be taken up and used by neighbouring cells. Although lentivirus-based strategies are safer than retrovirus approaches, concerns about potential insertional mutagenesis still exist.76
A first-in-human Phase I clinical study (NCT02800070) is testing autologous lentivirus-transduced HSPCs injection in male patients with classical FD.76 In 1 week, all patients had normalised production of α-Gal A. To date, no adverse effects have been reported. The first attempts at in vivo gene therapy used adenovirus because of its versatility.77 However, toxicity reports led to the withdrawal of this approach. Hepatocellular necrosis emerged as a relevant drawback. Indeed, the hepatotropic nature of the vectors used led to an inflammatory response that triggered both innate and adaptive immunity. As a result, transduced cells were targeted and destroyed.78 Thus, the focus has moved toward the adeno-associated virus (AAV).
Four AAV experimental therapies are being studied: FLT-190, ST-920, AMT-191 and 4D-310. Preclinical data on FLT190 showed safety and a steady rise in α-Gal A activity in mice and non-human primate models.79 These results led to the Phase I/II MARVEL-1 study (NCT04040049). Untreated and previously treated adult male patients with classic FD were recruited. After hopeful results from the first dosage group, in which the medication was well tolerated and provided a persistent and dose-dependent increase in α-Gal A plasma level, the study is now recruiting patients for the second dose group.
A Phase I/II dose-ranging multicentre study, STAAR (NCT04046224), is evaluating isaralgagene civaparvovec (ST-920) in adult patients. Five patients were on ERT at the beginning of the study. Four of them managed to cease the enzyme infusions without a relapse in Gb3 levels. AMT-191 increases the concentration of N-acetylgalactosaminidase (NagA). This enzyme enables Gb3 clearance while escaping from the antibody response. Finally, despite the potential additional benefits of a cardiac-directed capsid, three patients treated with 4D-310 developed atypical haemolytic-uraemic syndrome. Therefore, 4D Molecular Therapeutics ceased recruitment.
The potential benefit of gene therapies should be evaluated in the context of issues and concerns regarding current gene-editing techniques. Safety is still the major concern in gene editing given its potential to produce off-target effects, which could lead to severe genotoxic effects. Moreover, the cost of treatment is expected to be extremely high. However, it represents a unique opportunity to provide long-term treatment of FD, thereby removing the dependence on ERT or chaperone therapy.
Conclusion
The importance of early suspicion and diagnosis, multidisciplinary evaluation and management, and targeted therapy in FD demonstrates the need for precision medicine, from diagnosis to therapy, in this rare disease. Future developments are expected in preclinical diagnosis with novel biomarkers and effective therapeutic approaches to each stage of the disease. 