










Sarcoidosis is an idiopathic, heterogeneous systemic condition characterised by non-necrotising granulomatous inflammation, most commonly affecting the lungs and thoracic lymph nodes. Cardiac sarcoidosis (CS) may accompany multisystem manifestations or occur in isolation and it remains a diagnostic and therapeutic challenge, with a paucity of data to guide management. The natural history is not well known, and the presentation can vary from asymptomatic cardiac inflammation to high-grade atrioventricular (AV) block, ventricular arrhythmia, heart failure, and/or sudden cardiac death. Clinically manifest cardiac involvement is estimated to occur in approximately 25–50% of cases of systemic sarcoidosis depending on the means of ascertainment.1–3 Cardiac involvement conveys a worse prognosis and a recent large contemporary registry demonstrated a higher 10-year risk of heart failure, arrhythmic complications, and all-cause mortality in patients with sarcoidosis compared with matched control subjects.4
Despite a limited evidence base, corticosteroids remain the cornerstone of treatment for active CS. Limited data exist on who and when to treat, and the optimal immunosuppression regimen and duration of therapy remains unknown. This review aims to describe the indications for immunosuppressive therapy in CS and the commonly used therapeutic agents and combinations, and will present a centre-specific, interdisciplinary approach to treatment planning and the monitoring of therapeutic response.
Immunopathogenesis of Cardiac Sarcoidosis
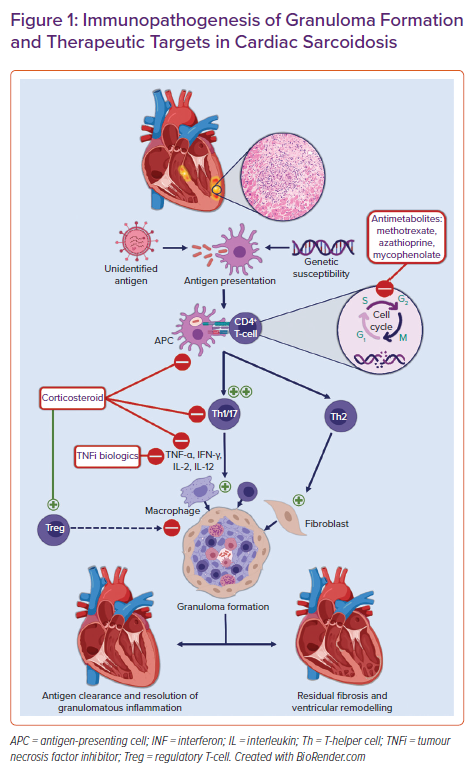
Non-necrotising granulomas are the histopathological hallmark of sarcoidosis. Granulomas are organised collections of macrophages and epithelioid cells surrounded by lymphocytes, fibroblasts, and collagen. The inciting precipitant for their formation in CS is not known. Multiple hypotheses about the trigger for granuloma formation abound. These range from infectious agents to environmental particles to an autoantigen with a dysregulated antigenic or T-cell response in individuals with a potential genetic predisposition and/or certain human leucocyte antigen (HLA) polymorphisms.5,6 Antigen-presenting cells, such as macrophages and dendritic cells, process this antigen and activate naïve CD4 T-cells, resulting in the proliferation of exaggerated T-helper (Th)1 and Th17 T-cell populations. These cells promote cell-mediated immunity and secrete pro-inflammatory cytokines such as interleukin (IL)-2, IL-12, tumour necrosis factor (TNF)-α, and interferon (IFN)-γ.7 These cytokines aggregate macrophages, giant cells, and lymphocytes into tightly packed granulomas surrounding the inciting antigen. In the chronic phase, there is a shift from Th1 to Th2 T-cells secreting various cytokines and chemokines including IL-4, IL-10, and tumour growth factor (TGF)-β. These cytokines promote fibroblast recruitment and extracellular matrix deposition and fibrosis.5,8 Compounding this aberrant pro-inflammatory response is the decreased efficacy of regulatory T-cells (Tregs). Tregs normally maintain immune homeostasis and prevent autoimmunity by suppressing granuloma formation.9,10 An overview of the proposed pathogenesis of granulomatous inflammation and associated treatment targets in CS are shown in Figure 1.
Diagnosis of Cardiac Sarcoidosis
The clinical presentation of CS ranges widely, from an incidentally discovered condition to heart failure, bradyarrhythmias and tachyarrhythmias, or even sudden death. Diagnosing CS can be challenging given the lack of a single blood or imaging biomarker and the paucity of prospective studies centred on diagnostic criteria. As a result, CS may be under-recognised in clinical practice and an appropriate index of suspicion is required during diagnostic evaluation.
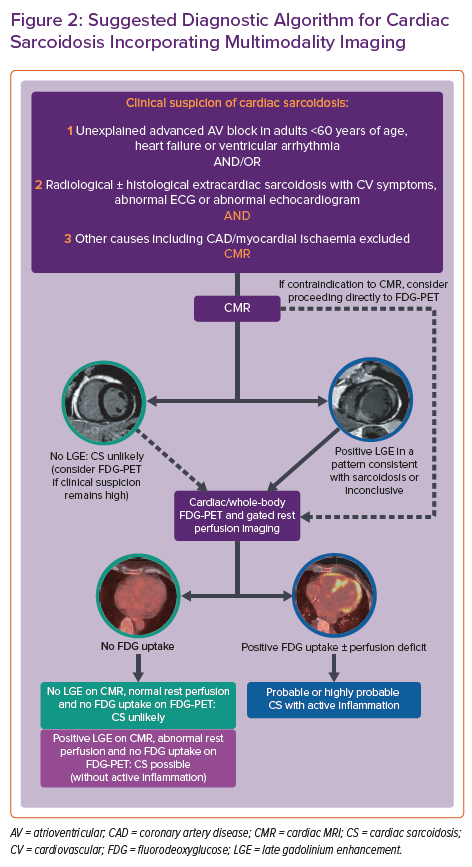
In 2014, the Heart Rhythm Society (HRS) developed an expert consensus statement to provide standardised contemporary recommendations on the diagnosis and treatment of CS.11 These diagnostic criteria incorporate either a histological diagnosis from myocardial sampling or a clinical diagnosis based on appropriate clinical context and cardiac imaging, when there is biopsy-proven extracardiac sarcoid. Given the heterogeneous presentation and often elusive nature on imaging or histological sampling, the probability of a diagnosis of CS can be determined using the World Association for Sarcoidosis and Other Granulomatous Disorders (WASOG) organ assessment instrument, which defines three categories of probability of organ involvement in sarcoidosis: highly probable (>90% likelihood), probable (50–90% likelihood) and possible (<50% likelihood of organ involvement).12 A probable diagnosis is considered adequate to establish a clinical diagnosis of CS according to the expert consensus recommendations.11 When immunosuppressive therapy is being considered in suspected CS, a complementary imaging approach involving cardiac MRI (CMR) and fluorodeoxyglucose-PET (FDG-PET) is often used to determine the likelihood of the diagnosis and improve diagnostic accuracy.13 When FDG-PET information is added to CMR, up to 45% of patients are reclassified as having a higher or lower likelihood of CS.14 Both modalities have also been shown to provide independent prognostic information in suspected CS.15–18 A suggested diagnostic algorithm using multimodality cardiac imaging and incorporating the current HRS and WASOG consensus statements is shown in Figure 2.11,12,14
Isolated Cardiac Sarcoidosis
Isolated CS may account for up to 25% of cases of CS.19 Diagnostic certainty for this entity is often limited and the current diagnostic criteria for CS have limited sensitivity given the absence of extracardiac disease and the low sensitivity of right ventricular endomyocardial biopsy.11,14,15 A combined approach to diagnosis using CMR and FDG-PET is often required to more accurately determine probability. Isolated CS has been associated with worse prognosis compared with systemic sarcoidosis and cardiac involvement.20
Mimics of Cardiac Sarcoidosis
Potential mimics include lymphocytic myocarditis, hibernating myocardium due to underlying obstructive coronary artery disease, some genetic cardiomyopathies, and physiological FDG uptake in heart failure. A high index of suspicion is required for giant cell myocarditis (GCM), although compared with CS this idiopathic inflammatory condition is typically marked by a heavier burden of arrhythmia and haemodynamic compromise. CS and GCM may even present as a continuum of a single inflammatory process rather than as two distinct entities. Arrhythmogenic cardiomyopathy due to desmoplakin mutations can mimic active isolated CS and present with episodic acute left ventricular (LV) myocardial injury and associated myocardial inflammation on FDG-PET.21 As a result, three-generation pedigree and genetic testing should be undertaken in selected cases when familial cardiomyopathy is suspected prior to embarking on immunosuppressive therapy.
Indications for Immunosuppression
Much of how myocardial inflammation in CS is treated is based on limited retrospective evidence or extrapolation from studies of extracardiac disease. The HRS consensus statement makes recommendations for immunosuppression in the setting of myocardial inflammation by either myocardial histology, CMR or FDG-PET in the following scenarios: high-grade AV block; frequent ventricular ectopy or non-sustained ventricular tachycardia (VT) and evidence of myocardial inflammation; and sustained ventricular arrhythmias and evidence of myocardial inflammation.11 Although no formal recommendation was made on the use of immunosuppressive therapy in patients with LV systolic dysfunction and myocardial inflammation, this is a widely accepted indication for consideration of immunosuppressive therapy in CS.22
The goal of therapy is to reduce myocardial inflammation to limit the development of myocardial fibrosis and the associated re-entrant ventricular arrhythmias, heart block, and new or worsening of LV dysfunction and heart failure. Some also hypothesise that active inflammation itself may be a source of ventricular arrhythmias in CS. The presence of FDG uptake in addition to abnormal perfusion in patients with suspected CS is associated with a higher risk of death or sustained VT even when adjusted for LV ejection fraction (LVEF). However, it is not known if a reduction in the extent of inflammation translates to a reduction in adverse clinical events.15 Patients with right ventricular inflammation should also be considered for treatment, given an association with worse prognosis.15,23
Whether immunosuppressive therapy should be initiated in patients with asymptomatic metabolically active CS on FDG-PET and normal ventricular function is less clear and is largely extrapolated from studies of those with LV dysfunction. In these cases, individualised assessment is required, including balancing the amount of myocardial inflammation, the extent of systemic involvement, and the clinical likelihood of CS (if uncertain) with the potential risks of therapy. In such cases, the presence of both a resting myocardial perfusion defect and FDG uptake on FDG-PET may be useful in the decision-making process given their association with adverse cardiac outcomes.15,23
Conduction Disease
Sarcoidosis may affect any component of the cardiac conduction system but symptomatic high-grade second-degree or third-degree AV block is the most common presenting feature of CS, with an incidence of 42% from Finnish registry data.24,25 High-grade AV block in CS is thought to occur due to involvement of the basal interventricular septum by either granulomatous inflammation or residual fibrosis. Immunosuppressive therapy should be considered in the setting of advanced AV block.
Although the evidence base for immunosuppressive therapy in CS is limited, much of it focuses on the effect of corticosteroids on the recovery of AV nodal conduction. In the absence of prospective randomised data in this area, a 2013 meta-analysis examined 10 studies of poor to fair quality in 299 patients with CS.26 This included 73 patients with AV conduction disease, 57 (78%) of whom were treated with corticosteroid. Of those treated, 47.4% had improved AV conduction while none of the 16 patients who did not receive corticosteroid therapy improved. Variable definitions were used for AV conduction disease and recovery in these studies. However, most focused on third-degree AV block that resolved with treatment.
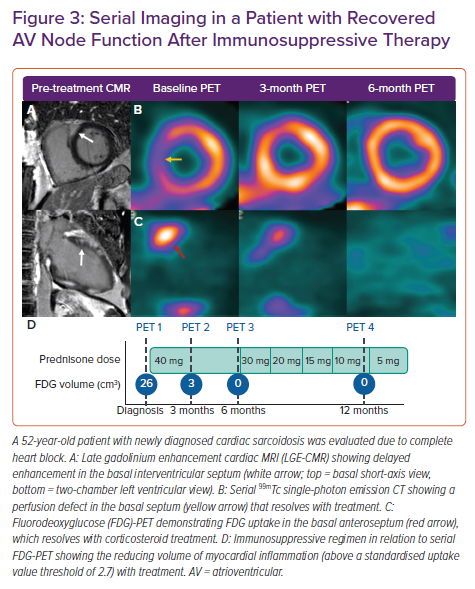
Imaging of myocardial inflammation in the region of the AV node with either FDG-PET or T2-weighted CMR has the potential to predict recovery of AV nodal function and guide immunosuppression. A retrospective study examined response to corticosteroids in 10 patients with newly diagnosed CS and third-degree AV block who had undergone both CMR with late gadolinium enhancement (LGE) and FDG-PET.27 All patients had pre-treatment LGE in the basal interventricular septum. The six patients with functional AV nodal recovery had pre-treatment focal inflammation in the basal interventricular septum on FDG-PET, while those without recovery had either no FDG uptake or FDG uptake with thinning of the interventricular septum. Although the number of subjects was small, this suggests that AV nodal recovery after steroid therapy may be more likely in those with basal septal inflammation on metabolic imaging but with preserved wall thickness, and that recovery may be possible even in the presence of LGE in the region of the AV node (Figure 3).
Although AV conduction disease may resolve with corticosteroid treatment, isolated AV block due to CS is not a benign entity. A retrospective study from Japan that included 22 patients with high-grade AV block found that 41% had a fatal major adverse cardiac event over a median of 34 months, principally due to ventricular arrhythmia.28 Similar fatal event rates were seen in the subgroup who recovered normal AV nodal function after the initiation of corticosteroid therapy. This reinforces the consensus recommendation for ICD implantation whenever permanent pacing is required in CS, given that the potential for reversibility is unpredictable and even with reversibility, the risk of sudden cardiac death persists.11 It is important that appropriate cardiovascular implantable electronic device (CIED) therapy should not be delayed while awaiting response to immunosuppressive therapy.
Ventricular Arrhythmias
Ventricular arrhythmia is a feared complication of CS and a significant predictor of mortality.29 The predominant mechanisms of arrhythmogenesis are abnormal automaticity and triggered activity in the inflammatory phase and scar-mediated re-entrant circuits in areas of residual fibrosis.30,31 Multiple VT mechanisms can be present in the same patient depending on disease stage. An FDG-PET-based study in patients with CS found that myocardial inflammation intensity was greater and LVEF lower in those presenting with VT compared with those with AV block or controls with clinically silent CS.32
The evidence base for immunosuppression in patients with ventricular arrhythmias and myocardial inflammation is limited and no randomised data exist. However, the current expert consensus recommendation is that immunosuppression and anti-arrhythmic drug therapy should be administered if active inflammation is present and should occur in tandem with defibrillator implantation when appropriate.33 These measures may be considered over catheter ablation when active inflammation is present in the setting of VT.31 Conversely, in CS patients without active inflammation, immunosuppressive therapy as a component of VT management is generally not indicated.
In a retrospective study of corticosteroid treatment for frequent premature ventricular contractions (PVCs) in 31 patients with CS, there was no difference in PVC burden or prevalence of non-sustained VT before and after steroid treatment.34 However, when stratified by LV systolic function, these events were significantly reduced in the cohort with LVEF ≥35%. Moreover, this subgroup contained all patients in the study with uptake on gallium scintigraphy, indicative of myocardial inflammation. Resolution of uptake occurred in 80% of these patients, suggesting greater benefit of immunosuppressive therapy in ventricular arrhythmia before the onset of a more scarred, remodelled ventricle and in the presence of active inflammation. In another retrospective study, 14 patients with CS presenting with VT and PET-FDG uptake had a good early response to a combination of prednisolone and methotrexate (13/14).35 Four patients had recurrence of VT during follow-up, three of whom had disease reactivation on PET, all of whom had no further VT with intensified immunosuppression.
In cases of ventricular arrhythmia, the presence and extent of myocardial inflammation should ideally be defined with either FDG-PET (or T2-weighted CMR) before commencing immunosuppressive treatment. However, in certain cases of ventricular arrhythmia in patients with known CS who may be too unstable to undergo advanced imaging, empirical treatment is recommended.11
Left Ventricular Dysfunction
Symptomatic heart failure as the initial manifestation of cardiac involvement in sarcoidosis is less common than either high-grade AV block or ventricular arrhythmias (18% in Finnish Registry).20 LV systolic dysfunction at presentation has been well described as an independent predictor of adverse outcomes and mortality irrespective of the degree of myocardial inflammation, with a 10-year transplantation-free survival of only 53%.20,26,29
Chiu et al. investigated the effect of corticosteroid therapy over a mean follow-up of 88 months in the prevention of LV remodelling.36 Forty-three patients treated with prednisolone were retrospectively stratified by LVEF. Those with an LVEF >55% had unchanged LV volumes and function after treatment while those with an LVEF 30–55% had a significant reduction in volume and improvement in LVEF. Patients with an LVEF <30% had no positive remodelling or improvement in LVEF (LVEF decreased from 22% to 19%; p=0.08). Pre-treatment decreased LVEF was associated with increased mortality. Nagai et al. found that a cohort of patients treated with steroids, the majority of whom had myocardial inflammation identified on either Gallium scintigraphy or FDG-PET, had a greater increase in LVEF and reduced heart failure hospitalisations but no difference in cardiac death or arrhythmias compared with those who did not receive steroids.37 These studies support the role of corticosteroids in preventing and reversing LV remodelling in the early and middle stages of the disease, and suggest that immunosuppression may not be as effective in later stages when fibrosis predominates and more advanced LV dysfunction is present.
Therapeutic Agents
Corticosteroids
Oral corticosteroids are commonly used as first-line therapy for active CS and are the focus of the majority of available research in this area.11,26,38 Steroids non-selectively suppress production of the cytokines involved in granuloma formation including TNF-α and IFN-γ (Figure 1), inhibiting inflammatory cell migration and restoring CD4+ T-cell function as well as the balance between the subtypes of effector CD4+ T-cells seen in sarcoidosis.7,10
The optimal dose, duration, and tapering regimen for corticosteroid therapy have not been established. The Japanese Circulation Society recommended an initial prednisolone dose of 30 mg daily or 60 mg on alternate days for a 4-week period, followed by tapering of 5 mg monthly to reach a maintenance dose of 5–10 mg daily or 10–20 mg on alternate days by 6 months.38 However, the extent of myocardial inflammation and severity of clinical presentation may warrant a higher starting dose. Although oral steroids are most commonly used, small case series have described the use of IV methylprednisolone at doses of 500–1,000 mg for 1–3 days in the acute treatment of refractory ventricular arrhythmias in the setting of active myocardial inflammation.39,40
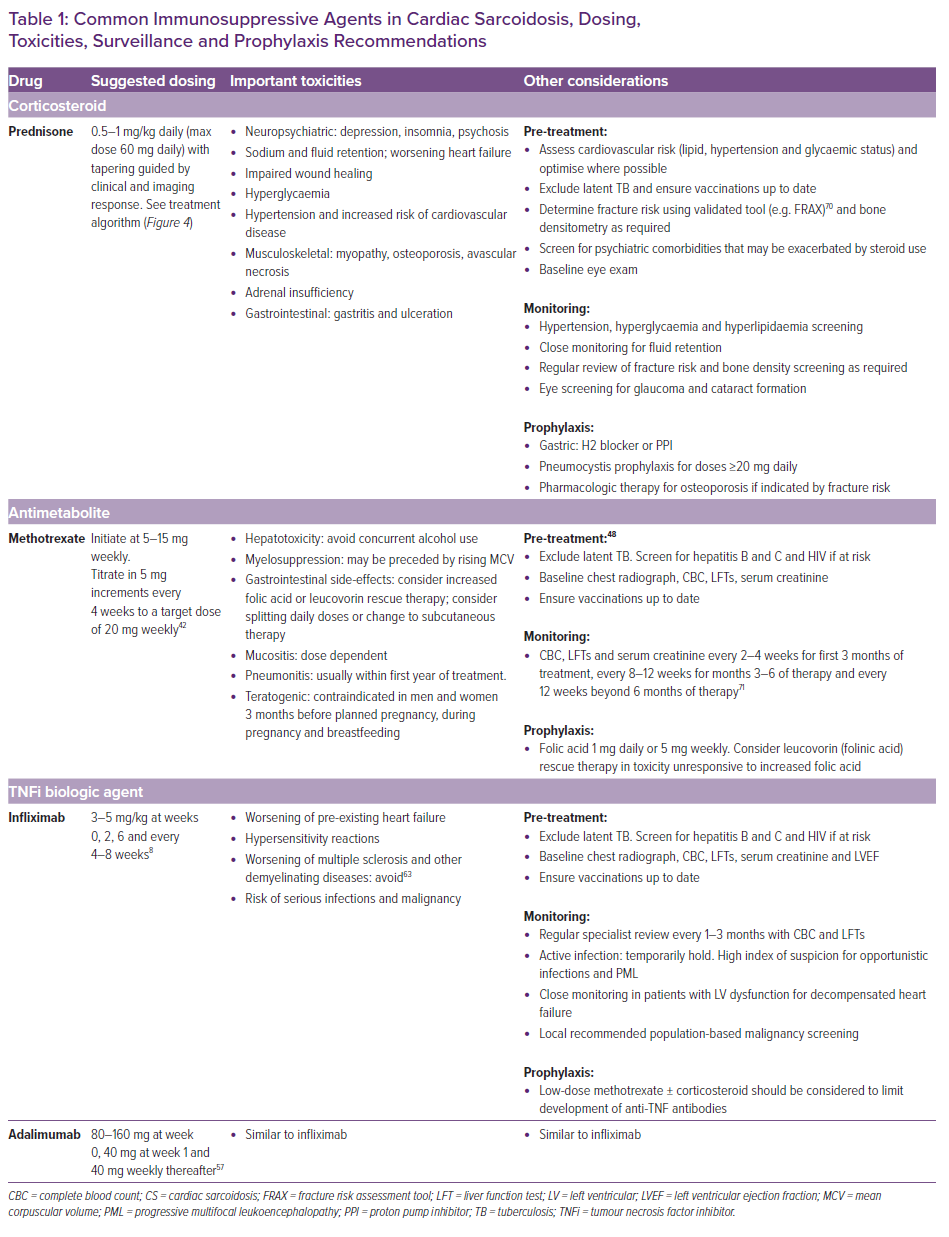
A prolonged need for immunosuppressive therapy is often required and therefore early addition of a steroid-sparing agent is frequently used to minimise steroid toxicity while achieving early remission of inflammation. The median duration of steroid treatment in a larger retrospective cohort with CS was 22 months, although in that study the steroid treatment was not associated with improved outcomes.41 It is unknown what the risk of disease relapse is following steroid cessation when treatment is guided by FDG-PET. The baseline potential for corticosteroid toxicity should be factored into the decision-making process prior to initiation. Important toxicities that should be anticipated and which are recommended for monitoring are listed in Table 1.
Methotrexate
Methotrexate is recommended as the second-line or add-on therapy for systemic sarcoidosis in the case of inadequate response to steroid therapy or to minimise cumulative steroid exposure.42 It may be used in the first line as a methotrexate–steroid combination or as monotherapy in cases when exposure to corticosteroid is best avoided. Methotrexate regulates the cellular function of multiple cells involved in inflammatory pathways including T-cells (Figure 1). Methotrexate inhibits the folate-dependent de novo synthesis of purines and pyrimidines necessary for inflammatory cellular replication. It also increases the extracellular adenosine level, which may have anti-inflammatory effects.43 Methotrexate is the most widely used antimetabolite or steroid-sparing agent in pulmonary sarcoidosis and has been extrapolated to the treatment of CS.44
Baughman et al. completed a small randomised controlled trial of patients with acute pulmonary sarcoidosis who took 10 mg methotrexate weekly versus placebo and found that the methotrexate group required a significantly lower dose of corticosteroid at 12 months.45 A prospective study that compared 17 patients with CS treated with either a combination of methotrexate and low-dose steroid (5–15 mg/day) or steroid monotherapy found that at 3 years, LVEF and N-terminal pro-B-type natriuretic peptide were significantly more stable in the combination group.46 Methotrexate added to low-dose prednisone (<10 mg/day) after 4–8 weeks of high-dose steroid has also been shown to have a high rate of remission of inflammation on FDG-PET, supporting its use as a steroid sparing agent.47
Methotrexate should be dose adjusted in the case of renal impairment (50% dose reduction for estimated glomerular filtration rate [eGFR] 30–49 ml/min/1.73 m2) and avoided in advanced renal impairment (eGFR <30 ml/min/1.73 m2) or non-sarcoidosis-related hepatic dysfunction.48 A starting dose of 5–15 mg weekly is recommended, followed by uptitration by 5 mg every 2–4 weeks to a target of 20 mg with serial monitoring for leukopenia, renal dysfunction, and hepatotoxicity (Table 1).42 Steady-state levels of methotrexate and maximum therapeutic effect can take up to 6 months to become apparent, which should be factored into the surveillance plan for FDG-PET if monotherapy is used.49 The most frequent adverse events are fatigue and gastrointestinal side-effects, while the incidence of clinically important cytopenia is estimated to be <1%.49 Concurrent folic acid should be given at a dose of 1 mg daily or 5 mg weekly to minimise fatigue, gastrointestinal side-effects and hepatotoxicity without compromising efficacy.50 Higher dose folic acid or a transition to leucovorin (folinic acid) can be used as rescue therapy to minimise these side-effects. Longer term use of methotrexate for up to 2 years has been shown to be well tolerated in patients with systemic sarcoidosis at a mean dose of 10 mg weekly.51
Alternative Steroid-sparing Agents
Azathioprine, mycophenolate, leflunomide, hydroxychloroquine, cyclophosphamide, and sirolimus are alternative steroid-sparing agents that have been used in extracardiac sarcoidosis in those unresponsive to or intolerant of methotrexate. However, their use in CS is limited to case reports. Fussner et al. reported on their longitudinal experiences of management of CS across two large North American centres and noted that mycophenolate mofetil was their most commonly used steroid-sparing agent (37%), but limited data are available to support its use over other agents.41
Biologic Therapy
Patients with inflammatory disease refractory to tiered therapy with corticosteroid and a steroid-sparing agent such as methotrexate should be considered for third-line treatment with a biologic agent, such as a TNF-α inhibitor. In one large retrospective cohort with CS, biologic therapy was used in 8%.41 TNF is a cytokine central to the development and maintenance of granulomas, and TNF-α inhibitors including infliximab (chimeric monoclonal antibody) and adalimumab (humanised monoclonal antibody) have been used in CS. Infliximab has been used with good effect in refractory pulmonary sarcoidosis, albeit with substantial adverse event and discontinuation rates of up of 52% and 23%, respectively, in one study.52–54
A retrospective study by Harper et al. of 36 patients with refractory CS treated with infliximab demonstrated a significant reduction in steroid dosing at 6 and 12 months without worsening of heart failure.55 Both infliximab and adalimumab have been shown to reduce the extent and intensity of inflammation on serial FDG-PET in a multicentre retrospective study with no significant change in LVEF, with 11% requiring hospitalisation for HF and a 5% discontinuation rate.56 The optimal duration for TNF-α inhibitor therapy and the risk of disease relapse in CS is not known, but expert consensus recommends continuing for at least 3–6 months, with response most frequently observed in the first month of treatment when these agents are used in rheumatic inflammatory conditions.57
Due to the potential for the development of neutralising antibodies to biologic therapy that can limit efficacy, concurrent treatment with methotrexate (typically 10 mg weekly) is often pursued to mitigate this risk.58
Alternative TNF-α inhibitors golimumab and etanercept have not been found to be effective in extracardiac sarcoidosis.8 Rituximab, an antiCD20 B-cell-depleting humanised monoclonal antibody, has shown limited results in a small prospective study of refractory pulmonary sarcoidosis, while its effectiveness in CS is limited to case reports.59,60
Importantly, infliximab at a dose of 10 mg/kg was associated with an increased risk of worsening heart failure or death in a randomised trial conducted in patients with reduced LVEF and New York Heart Association (NYHA) class III–IV heart failure. Although that study was not conducted in patients with CS per se, we generally avoid TNF-α inhibitors in patients with severe systolic dysfunction and associated heart failure for this reason.61,62
Biological therapy should not be initiated in the presence of serious active infection, and screening should be performed for tuberculosis before initiating treatment, as well as for viral hepatitis and HIV in those at risk.63 These agents should not be commenced in patients with a known or suspected malignancy. Although there is no conclusive evidence for an increased risk of lymphoproliferative disease or solid organ malignancy with TNF-α inhibitor treatment in patients with rheumatological conditions, vigilance is recommended, with standard screening and preventive measures.63
Monitoring Response to Treatment
Clinical assessment, device interrogation, and echocardiography are used routinely in the longitudinal follow-up of patients receiving immunosuppressive therapy for CS. However, it is not possible to determine the presence of ongoing myocardial inflammation from these studies alone, or to differentiate between active inflammation and myocardial fibrosis as a cause of clinical events. To date, no cardiac-specific or inflammatory blood-based biomarkers have been found to correlate with the presence and extent of either cardiac or extracardiac granulomatous inflammation, nor have there been any studies on their use in guiding immunosuppressive therapy.
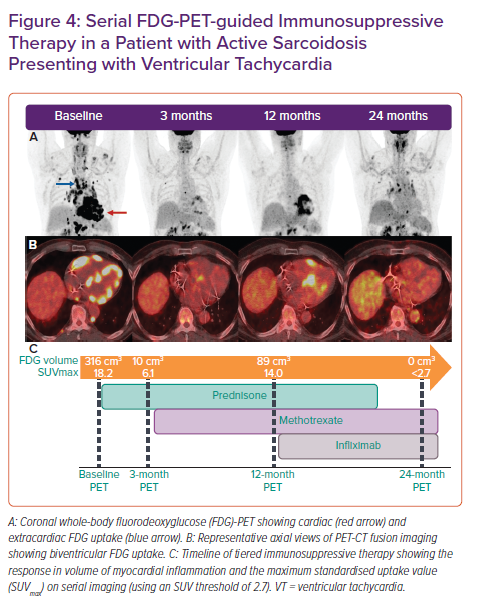
FGD-PET-CT is commonly integrated with echocardiography, CMR, and tissue sampling for the diagnosis and monitoring of response to immunosuppression in CS (Figure 4). Although there is a growing evidence base supporting the role of FDG-PET in the diagnosis and prognostication of CS, data on the optimal interval between follow-up assessments and on the potential for PET-guided therapy are limited.15,16 Whether resolution of myocardial FDG uptake is the optimal endpoint for immunosuppressive treatment in CS remains unclear.
At our institution, Osborne et al. retrospectively studied 23 patients with CS who were predominantly treated with corticosteroid and underwent serial FDG-PET with a median of four scans per patient.64 We demonstrated that a reduction in the intensity and extent of myocardial inflammation measured quantitatively (maximum standardised uptake value [SUVmax] and the volume of myocardial FDG uptake above a prespecified SUV threshold) was associated with an improvement in LVEF. These data are promising but it is not yet known whether this reduction or resolution of myocardial inflammation translates into a reduction in adverse clinical outcomes or residual myocardial fibrosis, and which treatment regimen, if any, serves best to achieve this inflammatory resolution. Persistence of perfusion defects on FDG-PET following steroid therapy has been associated with higher rates of adverse outcomes and so may be a potential treatment target on serial imaging.65
To facilitate accurate and reliable comparison between serial FDG-PET scans when monitoring therapy response, a consistent reproducible technique is required at an institutional level with particular attention paid to patient dietary preparation in order to achieve adequate suppression of physiological myocardial uptake, which can interfere with interpretation.66
Treatment Approach
The treatment of myocardial inflammation in CS is largely empirical and highly variable. A survey of 42 sarcoidosis experts in 2012 was unable to demonstrate a consensus on the optimal dose of prednisone, the use of steroid-sparing agents, or the duration of treatment, highlighting the uncertainty and divergence of opinion that exists in the field.67 The treatment approach should focus on treating granulomatous inflammation in the context of patient-specific comorbid conditions while anticipating and offsetting common toxicities.
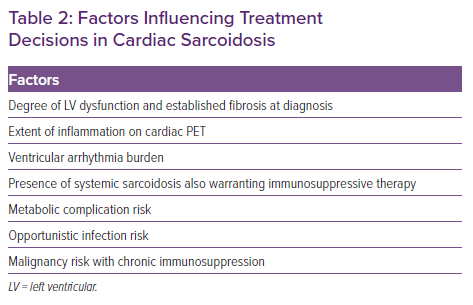
The treatment algorithm used in the Cardiac Sarcoidosis and Inflammatory Heart Disease Program at Brigham and Women’s Hospital, for those with clinically manifest CS and an accepted indication for immunosuppression (high-grade AV block, VT or heart failure) is shown in Figure 5. Our approach incorporates clinical status in combination with FDG uptake as the endpoint of therapy when possible. Immunosuppressive treatment in those with CS and isolated atrial arrhythmias or asymptomatic LV dysfunction is not represented in the currently available literature or expert consensus guidelines, and requires a patient-specific approach.
When anti-inflammatory therapy is indicated, a shared decision-making approach with the patient is used, taking into account several factors (Table 2). All patients commencing immunosuppressive therapy are screened for latent tuberculosis using a whole blood IFN-γ release assay (e.g. T-Spot). In the Cardiac Sarcoidosis and Inflammatory Heart Disease Program at Brigham and Women’s Hospital, we typically defer initiation of immunosuppressive therapy for 2–6 weeks after CIED implantation to allow for adequate wound healing and to minimise pocket infection risk.
For immunosuppressive therapy, we initiate oral prednisone at a dose of 0.5–1 mg/kg daily with a maximum daily dose of 60 mg. Depending on clinical response, we taper by 10 mg monthly to a target of 20 mg daily by the time of first follow-up FDG-PET scan at approximately 3–4 months. All patients receive pneumocystis pneumonia prophylaxis with either sulfamethoxazole/trimethoprim or atovaquone while requiring prednisone doses ≥20 mg daily, in addition to gastric ulceration and osteoporosis prophylaxis when appropriate (Table 1).
Given that overproduction of 1,25-dihydroxy vitamin D is common in sarcoidosis and can result in increased intestinal absorption of calcium, increased bone resorption and hypercalciuria with or without hypercalcemia, we avoid empirical initiation of calcium and vitamin D supplementation in patients with sarcoidosis.68 In general, we aim to wean the patients off corticosteroids over a 12–24-month period, as guided by clinical events and demonstration of resolution of myocardial inflammation on serial FDG-PET scans. This taper is often performed slowly at doses <20 mg daily so that any potential inflammatory reactivation can be detected early.
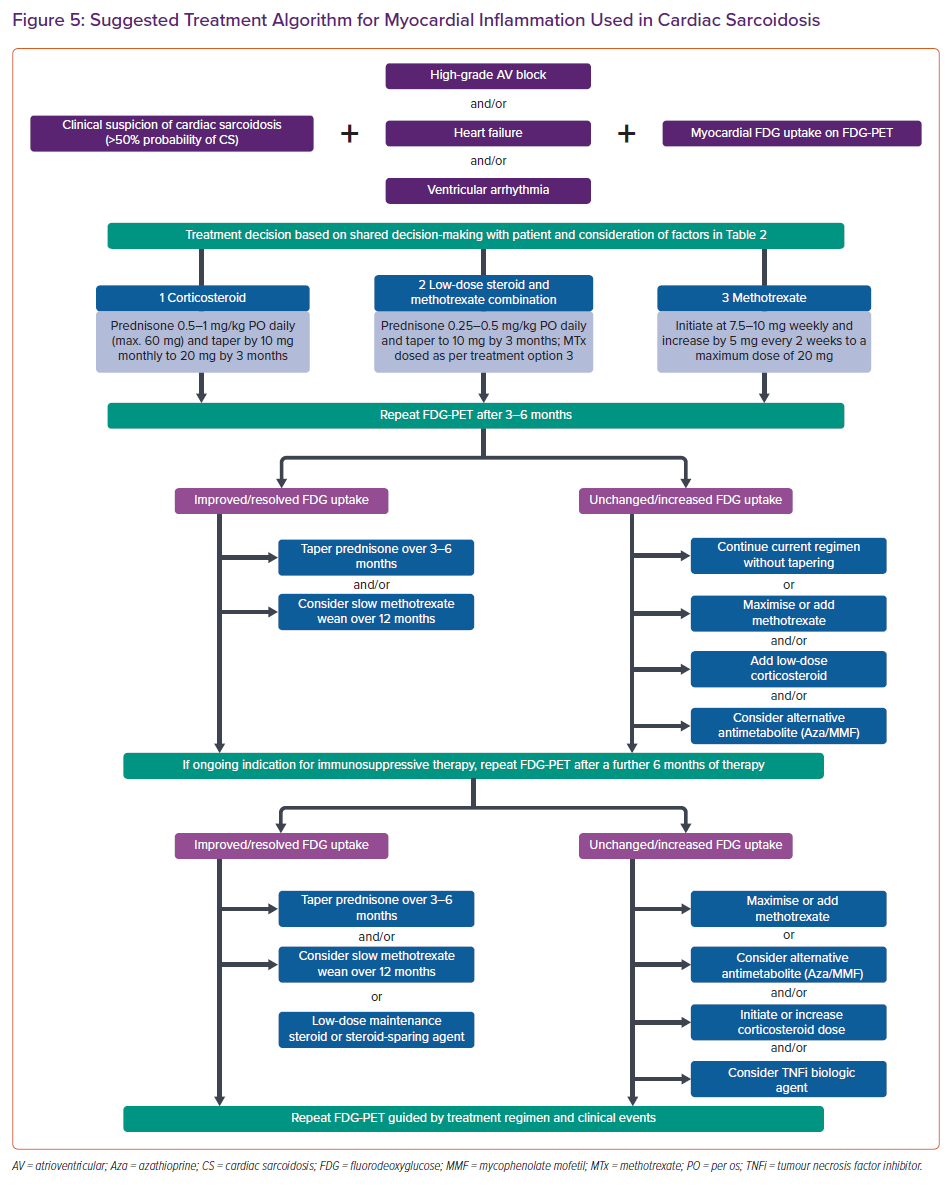
In patients with a contraindication to higher doses of corticosteroids, we typically initiate either methotrexate monotherapy or methotrexate as an adjunct to lower dose corticosteroid therapy. Methotrexate is initiated at a dose of 7.5–10 mg weekly. In general, this is titrated in increments of 2.5–5 mg weekly every 2–4 weeks to a target dose of 20 mg weekly, along with monitoring for leukopenia and hepatotoxicity (Table 1). If chronic maintenance methotrexate is required, we aim to use the lowest possible dose to suppress myocardial inflammation while minimising toxicity. In general, we use serial FDG-PET to guide treatment escalations and weaning, and for serial LVEF assessments (Figure 5). However, we recognise that the availability and cost of cardiac PET remain significant limitations to more widespread use. In these cases, we recommend a surveillance approach using a combination of serial echocardiography, ECG, and clinical assessments to monitor response to treatment. We use ECG to assess for recovery or progression of conduction system disease, and device interrogation for arrhythmia burden and recovery of AV nodal function (via the percentage of ventricular pacing) in those patients with high-grade AV block and a pacer. In patients who have an unsatisfactory response to tiered therapy with corticosteroid and a steroid-sparing antimetabolite agent, third-line treatment involves the addition of a biologic such as the TNF-α inhibitor infliximab in the absence of symptomatic heart failure with reduced LVEF.
For centres aiming to establish a dedicated CS clinic or service, we suggest the adoption of a diagnostic and treatment algorithm (Figures 2 and 5, respectively). Timely integration of both high-quality CMR and FDG-PET (incorporating rest perfusion imaging) into these algorithms is paramount for accurate diagnosis, immunosuppression surveillance, and prompt device therapy. Organised multisystem protocols should encompass, for example, pulmonary or ophthalmological screening as necessary and establish pathways for the expert review of histology and management of biological therapy as required.
Uncertainties and Future Directions
The treatment of inflammation and its relationship to clinical outcomes in CS remain poorly defined. As a community, our challenge is to discern whom we should treat, when we should treat them, and how we should do it. Some of the key uncertainties that remain relate to the effect of corticosteroid therapy on clinical outcomes; the identification of biomarkers or imaging parameters that allow us to best predict and monitor response to therapy; which subgroups of patients are suitable for initiation of first-line steroid-sparing agents to minimise the metabolic disarray; the relative efficacy of different anti-inflammatory agents (steroid versus steroid-sparing; low-dose versus high-dose steroid); the place of watch and wait approaches to treatment in those patients with myocardial inflammation but normal cardiac function or clinically silent CS; and if and when biological therapy should be considered.
The relatively small patient numbers, delayed recognition, and the elusive nature of CS with regard to histological diagnosis have limited randomised trials and the observational study of treatment in this arena. The CHASM CS-RCT (NCT03593759) is an ongoing multicentre, open-label, randomised controlled trial to evaluate initial treatment strategies in immunosuppression-naïve patients with clinically active CS.69 As the first randomised controlled trial in CS, it aims to demonstrate the non-inferiority of a low-dose combination corticosteroid and methotrexate regimen to the intermediate corticosteroid dosing traditionally used in CS. For inclusion, patients must have at least one out of the classical CS triad of high-grade AV block, ventricular dysfunction or ventricular arrhythmia, in addition to both myocardial and regional lymph node FDG uptake on a recent FDG-PET and either histological correlation or CT chest with typical sarcoidosis features. The primary endpoint will be the summed perfusion rest score on myocardial perfusion imaging as a measure of myocardial scar over a 6-month period. Patients will be randomised to either a combination therapy arm with prednisone 20 mg/day for month one, 10 mg/day for month two and 5 mg/day for month three, in addition to methotrexate 10–20 mg weekly for 6 months or a corticosteroid monotherapy arm with prednisone 0.5 mg/kg/day for 6 months, with a maximum dose of 30 mg daily. The trial has limitations, such as its relatively small, anticipated recruitment of 194 patients; the use of summed perfusion rest score as the primary endpoint to assess scar burden instead of myocardial FDG uptake as a measure of inflammation burden; and its open-label nature.
The MAGiC-ART trial (NCT 04017936) is currently evaluating the role of anakinra, an IL-1 blocker, in the treatment of active CS in a cohort of 28 patients using changes in C-reactive protein over a 28-day period as a primary endpoint and changes in FDG uptake on PET, changes in LGE on CMR and clinical events over the same time point as secondary endpoints.
While many unanswered questions abound in the treatment of patients with CS, the care of these complex patients often requires a collaborative effort by subspecialists across the spectrum of cardiovascular medicine and other multidisciplinary specialists at referral centres similar to the Cardiac Sarcoidosis Program at the Brigham and Women’s Hospital.