









Heart failure is a growing pandemic worldwide, and it is associated with a high burden of morbidity and mortality. Acute heart failure (AHF) is one of the most prevalent causes of adult patients’ hospitalisation. While the treatment of most cardiovascular diseases has significantly improved since the beginning of this century, the outcomes of AHF have not progressed significantly, and AHF still carries a substantial risk for in-hospital mortality, readmission and post-discharge mortality.1,2
The use of opioids to relieve breathlessness in patients with respiratory disease dates back to the late 19th century.3 The immediate relieving effect of morphine on the key symptomatic discomfort associated with AHF, dyspnoea, facilitated the categorisation of morphine as a beneficial treatment in this setting. The rationale for the use of morphine as a decongestive therapy was further supported by animal studies showing a certain shift of volume between central and peripheral circulation, and has been attributed as medical phlebotomy.4,5
Evidence supporting morphine treatment for reducing AHF-associated mortality or morbidity are lacking. Nevertheless, current guidelines and textbooks continue the historical tradition mentioned above, and still accept morphine as a viable option for treating AHF and accordingly it is commonly prescribed.6–8 In particular, the relief of dyspnoea and anxiety is generally considered beneficial and serves as a justification for morphine administration.
During the last decade, however, several retrospective studies raised concerns regarding the safety and efficacy of morphine in the setting of AHF.9–12 In the current review, the physiological effects of morphine on the cardiovascular and respiratory systems are summarised, as well as the potential clinical benefits and risks. This is followed by a discussion on the reported clinical outcomes associated with morphine treatment in AHF.
Effects of Morphine on the Cardiovascular System
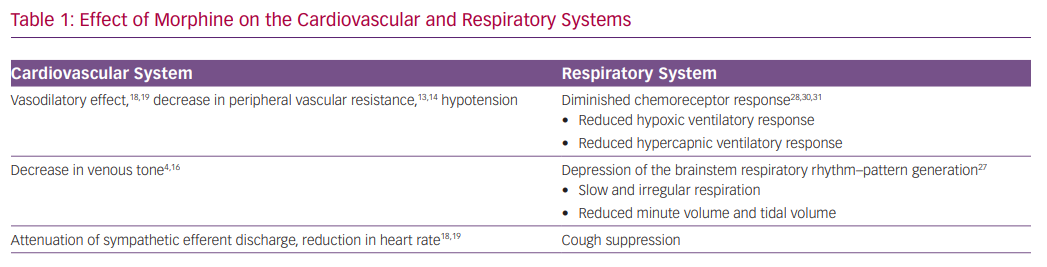
The effect of morphine on reducing vascular tone has been the key rationale for using the drug in the setting of AHF (Table 1). Animal studies have shown that morphine reduces both venous and arterial tone.4,5 Morphine exerts the vasodilatory effect mainly by an indirect increase in histamine release and not directly via the mu-opioid receptors.13,14 Henney et al. demonstrated that morphine administration at high doses (0.5–1 mg/kg) in dogs results in an immediate decrease in peripheral vascular resistance (46%) and in venous tone (49%). This was accompanied by an increase in venous capacitance of 11 ml/kg.4 The resulting increase in venous capacitance was presumed to be beneficial for patients with AHF by facilitating a shift of blood volume from the central to peripheral circulation. Other studies, however, identified only a minor effect on venous tone with a slight reduction in blood pressure.15,16
Zelis et al. studied 69 subjects who were treated with 15 mg of morphine.16 Venous pressure and tone decreased significantly by 34% and 38%, respectively. Mean systemic arterial pressure remained unchanged, while the authors reported a decrease of 47% in arteriolar constrictor response. A similar evaluation of patients with mild pulmonary oedema revealed a comparable reduction in venous tone that translated into a modest shift of 116 ml of blood to the peripheral circulation.17 Such a volume shift is too small to explain the presumed beneficial effect on congestion. Timmis et al. examined the effect of morphine in 10 patients with severe LV dysfunction following MI. In this study, morphine was associated with a significant and persistent reduction in heart rate and mean arterial pressure, and a small fall in cardiac index with minimal effect on systemic vascular resistance.18 The fall in cardiac output despite a reduction in systemic vascular resistance may be explained by attenuation of sympathetic efferent discharge from the central nervous system.16 Interestingly, Timmis et al. also observed a significant reduction in urine output, which was attributed to the stimulation of antidiuretic hormone release.18 In summary, there is little evidence, based on human studies, that morphine administration, in clinically relevant dosages, can exert a significant increase in venous capacitance that translates to the desired alleviation pulmonary congestion. Additional haemodynamic effects of morphine include decreases in heart rate and blood pressure.19
Effects of Morphine on the Respiratory System
There are substantial analogies between dyspnoea and pain, as both these subjective experiences involve several common cerebral structures.20 Opioids have powerful effects on respiration and its associated sensations, and their effectiveness in relieving dyspnoea is well-established.21 Opioids can also improve dyspnoea by reducing the associated anxiety.22
Morphine has a direct respiratory depressant effect by activating mu-opioid receptors in a dose-dependent manner.23 In knockout mice lacking mu-opioid receptors, administration of morphine and other opioids failed to induce respiratory depression.24,25 The mu-opioid receptors are expressed in areas of the central nervous system involved in respiratory rhythmogenesis and frequency control, particularly the pre-Bötzinger complex, but also in the cortex and the peripheral chemoreceptors.26–28
The central pattern generator of the respiratory neuron network extends from the facial nucleus to the spinal cord (the ventral respiratory group neuron network).29 The central pattern generator converts tonic excitatory chemo-drive into a respiratory pattern with distinct inspiratory and expiratory phases, and includes the parabrachial and Kölliker-Fuse nuclei in the pons, and the pre-Bötzinger and Bötzinger complexes.29 The pre-Bötzinger complex is the main region of respiratory rhythm–pattern generation, and is believed to be the main site of opioid-induced respiratory depression. In addition, opioids have profound effects on the cortical centres that control breathing, thereby augmenting their actions in the brainstem.27
The most opioid-sensitive aspect of respiration is rhythm generation, and changes in the respiratory pattern are observed at lower opioid doses than a change in tidal volume. Opioids cause respiration to slow and progress to become irregular (or cyclic) breathing, and eventually into apnoea.28 The induction of respiratory depression by opioids causes gradual hypercapnia that maintains respiration.
The peripheral chemoreceptors are responsible for 20–30% of the ventilatory drive at rest and for >80% during hypoxia, and express mu-opioid receptors.28 Consequently, opioids profoundly depress the hypoxic ventilatory response and the hypercapnic ventilatory response.28,30,31
Opioid-mediated respiratory depression has been studied mainly in the setting of acute and postoperative pain, with severe respiratory depression and related deaths occurring with an incidence of at least 0.5%.32,33 Data from studies conducted with patients experiencing chronic breathlessness demonstrated that opioid therapy (mostly oral) resulted in a small (~2 mmHg) increase in the levels of carbon dioxide without a significant effect on oxygen tension or saturation.34 While the risk for significant respiratory depression may be low in these settings, the associated hypoxia and the increased respiratory effort with pulmonary oedema is a vital compensatory mechanism for the compromised alveolar–capillary diffusion.
Morphine results in a robust and instantaneous anxiolytic effect by activation of the delta and mu-opioid receptors in multiple brain regions, and by reciprocal interaction with the GABAergic system.35,36 Morphine administration may also lead to several additional significant adverse events, including over-sedation, delirium, cough depression, nausea and vomiting, and urinary retention.37–40
Potential Clinical Benefits of Morphine in the Treatment of Acute Heart Failure
The haemodynamic benefits frequently cited as the rationale for using morphine to treat AHF include the reduction of venous tone with pooling of blood in the systemic (in particular, venous) circulation, peripheral arteriolar dilatation and antisympathetic effects (Table 2).4,5,15,41 Morphine-induced reduction in venous tone is considered advantageous in the setting of pulmonary oedema, as it may result in a volume shift from central to peripheral circulation. The venodilatation results in reduced venous return to the right heart and reduced right ventricular output, allowing the failing left ventricle to operate at a lower filling pressure, but may also lead to decreased cardiac output and hypotension, particularly with concomitant pulmonary hypertension. This negative effect on cardiac output may be mitigated by the mild reduction in systemic vascular resistance, resulting in decreased afterload and preserved stroke index.18 However, as discussed earlier, there is little compelling evidence that morphine causes either clinically significant venous pooling in the systemic circulation or a meaningful reduction of left or right ventricular filling pressures.16–18,42,43
Potential Clinical Risks of Morphine in the Treatment of Acute Heart Failure
Opioids decrease both hypoxic and hypercapnic respiratory drive, and this effect is directly proportional to the opioid dose and its analgesic potency.44 Induction of sedation and respiratory depression are among the most serious complications of morphine therapy. Opioids are second in the classes of medications contributing to adverse event reporting for hospitalised patients, with sedation and respiratory depression being among the most commonly reported adverse effects.45
Unfortunately, safety data on morphine use in the setting of AHF have not been systematically collected. Available data suggest that bolus administration of opioids is more likely to cause more severe respiratory depression than gradual administration.46,47 Various patient groups may be at higher risk for respiratory compromise, including the morbidly obese, patients who suffer from sleep apnoea, patients with respiratory muscle exhaustion and fatigue, patients with neurological or neuromuscular impairment, and the elderly. In addition, drugs, such as propofol and midazolam, have additive or synergistic effects on respiration when combined with opioids.28 Patients with metabolic alkalosis, which often accompanies diuretic therapy and depresses the respiratory centres, may also be susceptible to morphine.
These deleterious respiratory effects are accompanied by the increased risk for nausea and vomiting, consequently leading to further sympathetic activation, hindering the use of non-invasive ventilation, and augmenting the risk for aspiration and further respiratory compromise.48,49 The frequency of these dose-dependent complications were not reported in the setting of pulmonary oedema, but based on surgical series, the expected proportions of vomiting with 5 mg and 10 mg of morphine is 8% and 14%, respectively.50 The addition of 10 mg metoclopramide is recommended to counteract nausea if morphine is administered.49
Furthermore, morphine may affect the absorption of oral medications and therefore confer indirect harm. A randomised trial in the setting of acute MI recently demonstrated that morphine can delay clopidogrel absorption and decrease the plasma level of its active metabolite.51 A similar study revealed that the same effect is also relevant for ticagrelor administration.52–54 Inhibition of oral drugs absorption may be directly relevant to the treatment of AHF in the setting of MI, but presumably may be also relevant to treatment with oral drugs for AHF (e.g. thiazides and neurohormonal inhibitors). Finally, the haemodynamic effects of morphine can, directly and indirectly, affect urine output and renal function, resulting in decreased urine output in the setting of AHF.18,28,55,56
Morphine Therapy and Clinical Outcomes in Acute Heart Failure
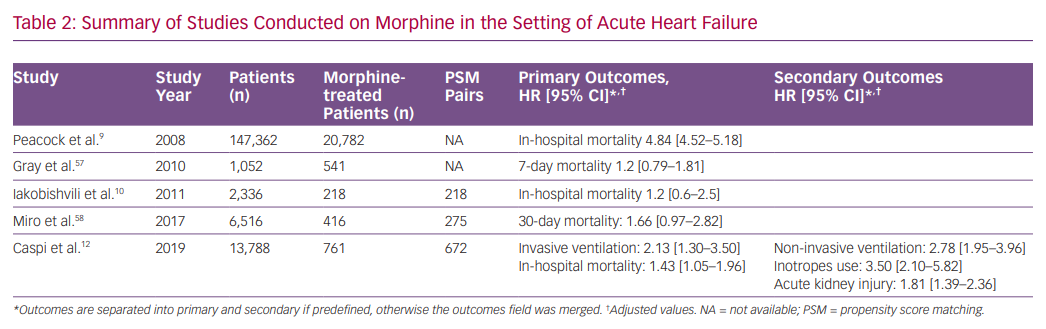
Morphine, together with nitrates and diuretics, is one of the most prescribed drugs for AHF, historically. Retrospective studies from the last decade raised doubts regarding the efficacy and safety of morphine therapy in AHF. In a large retrospective study including patients from the Acute Decompensated HEart Failure National REgistry (ADHERE), morphine therapy was associated with a marked increase in in-hospital mortality (OR 4.8), a higher rate of mechanical ventilation, intensive care admissions and longer duration of hospital stay.9 However, morphine dosage and timing of administration were not reported.
A significant limitation of any non-randomised analysis of the effect of morphine in AHF is that morphine-treated patients represent a cohort with more severe illness and would have been predicted to have greater mortality. Therefore, more recent studies applied propensity score (PS) matching to partially address these limitations.
In contrast, a study based on an Israeli registry of AHF (with two-thirds of morphine-treated patients having an acute coronary syndrome) showed that, in a multivariate analysis, morphine was associated with increased in-hospital mortality, but after PS matching (218 pairs), this effect was rendered insignificant.10 In addition, an analysis from the Three Interventions in Cardiogenic Pulmonary Oedema (3CPO) trial did not identify a relationship between opiate administration and mortality. However, opiate administration was independently associated with less improvement in arterial pH and did not improve breathlessness.57 Recently, a study based on the Spanish Epidemiology of Acute Heart Failure in Emergency Department (EAHFE) registry, which included 275 PS matched pairs, reported that morphine therapy during emergency department stay was associated with increased 30-day mortality (HR 1.66).58
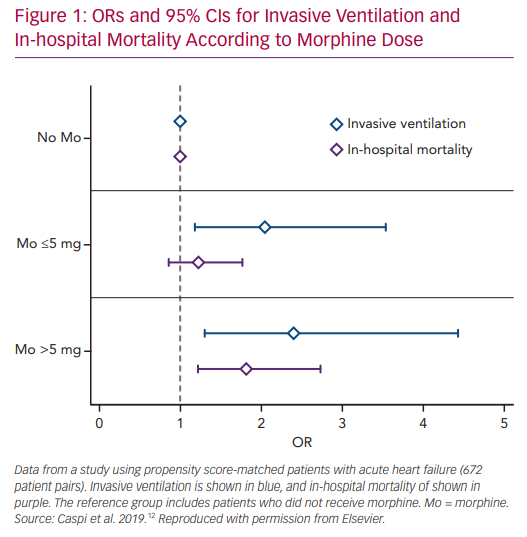
We recently studied the association between morphine use within the first 24 hours after admission and in-hospital clinical outcomes in 673 PS matched pairs of patients admitted with the primary diagnosis of AHF.58 Morphine therapy was associated with a significant increase in the need for subsequent invasive ventilation (OR 2.13; 95% CI [1.32–3.57], p=0.007) and in-hospital mortality (OR 1.43; 95% CI [1.05–1.98], p=0.024). Morphine therapy was also associated with a significant increase in the use of inotropes, non-invasive ventilation and acute kidney injury. Furthermore, we observed a significant direct relationship between morphine dose and the endpoints of invasive ventilation and mortality (Figure 1).
Importantly, we should approach the results of PS-based studies with caution. A full discussion on the merits and limitations of PS matching is beyond the scope of this review. Although PS matching provides excellent covariate balance, it frequently greatly reduced the sample size, resulting in a loss of both precision and generalisability.59 The fact that PS methods cannot control for unmeasured confounding is particularly relevant to studies on morphine use in patients with AHF.60 Such ‘unobserved confounders’ may be highly unbalanced in the treated and untreated groups, and may arise when clinicians use their expert knowledge (or gut feeling) to make the decision to administer morphine. Such clinical judgement is often based on unmeasured clinical characteristics that introduce significant bias.61 Finally, under some circumstances, PS matching may actually increase bias.60,62
Conclusion
Morphine administration in the setting of AHF is not currently encouraged by European guidelines (except for palliative and end-of-life care).7 Older US guidelines advocated morphine use in acute pulmonary oedema, but it is not mentioned in recent US guidelines.63,64 However, it is still frequently used, despite accumulating signals for harm.65 The continued use of morphine may be attributed to the desire to rapidly manage dyspnoea and anxiety per se, rather than waiting for these symptoms to improve with the resolution of the pulmonary oedema.
Some physicians believe that morphine-associated harmful effects may be restricted only to specific high-risk groups, such as those with hypoperfusion, low left ventricular ejection fraction or CO2 retention. However, the heterogeneity of treatment effect has not been shown in the above-mentioned studies.12,58 The association between morphine therapy and adverse events in AHF is complex, and likely requires the coexistence of several risk factors/patient susceptibilities to progress to a clinical event. Given the inability to acquire all the relevant patient data at the appropriate temporal resolution, causality remains to be established. However, there is little evidence that morphine is beneficial in the setting of AHF, and accumulating observational data demonstrating harm.
Clinicians encountering a distressed dyspnoeic AHF patient have no doubt that there is a compelling need for anxiolytic therapy, especially if this may be beneficial in alternative ways for the patient. Randomised controlled trials are required to assess the efficacy and safety of morphine in patients with AHF. In addition, the use of substituting agents, such as midazolam, may be explored. The ongoing MIdazolam Versus MOrphine in Acute Pulmonary Oedema (MIMO) randomised trial may provide this essential information.66 Alternative approaches focusing on agents with instantaneous anxiolytic effect and minimal respiratory depression (e.g. dexmedetomidine) may also be useful.67