







Central sleep apnoea (CSA) occurs in approximately one-third of patients with HF and is associated with a significant increase in morbidity and mortality compared to HF patients without CSA.1–3 CSA results in intermittent hypoxia and activation of the renin–angiotensin system, which contributes to worsening HF.4 Symptoms such as fatigue and difficulty concentrating often overlap with the effects of chronic HF. Treatment options are limited. Positive airway pressure (PAP) therapy has been the most commonly utilised but has failed to demonstrate improvements in quality of life (QOL) or HF.5,6 In addition, one form of PAP therapy increased mortality as a secondary endpoint in patients with reduced ejection fraction.6 A recently approved therapy has taken a physiological approach to treatment, stimulating the phrenic nerve to restore a normal breathing pattern.7 This therapy has been demonstrated to improve CSA and QOL.
Prevalence and Patient Presentation
The prevalence of CSA in patients with HF and reduced ejection fraction (HFrEF) is well documented and has remained remarkably stable, estimated at 37–51 %, even with guideline-directed medical therapy.1,8,9 There are fewer studies of CSA in people with HF and preserved ejection fraction, but the prevalence in this population remains approximately 30 %.10 The prevalence of CSA increases with the severity of HF, but it also occurs in patients with mild symptoms.4,9 CSA is also common in patients with other cardiovascular diseases; for example, it is found in approximately 31 % of patients with AF and normal LVF.11
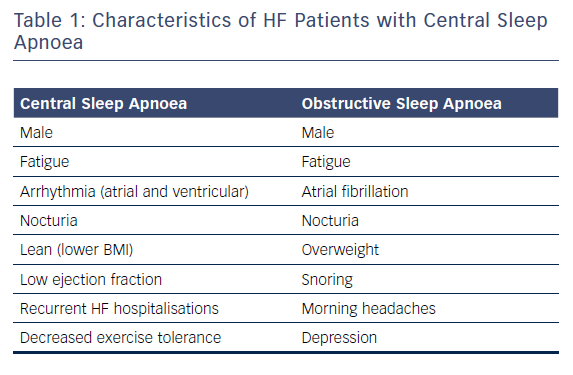
Patients with CSA and HF present with different symptoms from those typically seen in obstructive sleep apnoea (OSA), although some overlap exists (Table 1). HF patients with CSA tend to be thinner, older, have atrial arrhythmias and/or have daytime hypocapnoea (pCO2 <38 mmHg).12 Frequent symptoms include fatigue, insomnia and poor concentration. Patients may also report paroxysmal nocturnal dyspnoea, headache and nocturnal angina, which are thought to be due to intermittent hypoxia.4,12 Both OSA and CSA are more common in men, although it is unclear why this should be the case with CSA.4 Suspicion for sleep apnoeas should be high in the HF population. Regardless of the nature of the apnoea (either OSA or CSA), two-thirds of HF patients have sleep apnoea.
It seems reasonable to think that patients with CSA would be aware of their sleep disturbances, especially those in the severe category (apnoea–hypopnoea index [AHI] >30 events/hour). However, an interesting phenomenon in the HF population is that many patients do not recognise daytime sleepiness or report disrupted sleep, even in the presence of objective sleepiness.13,14 Multiple studies have shown a lack of daytime sleepiness in these populations. It has been theorised that this could be due to patients attributing their poor QOL and fatigue to their underlying HF.15 There is also a hypothesis that the increase in sympathetic drive in these patients causes them not to have typical ‘sleepiness’ symptoms, but rather results in a fatigued or hypervigilant state.16
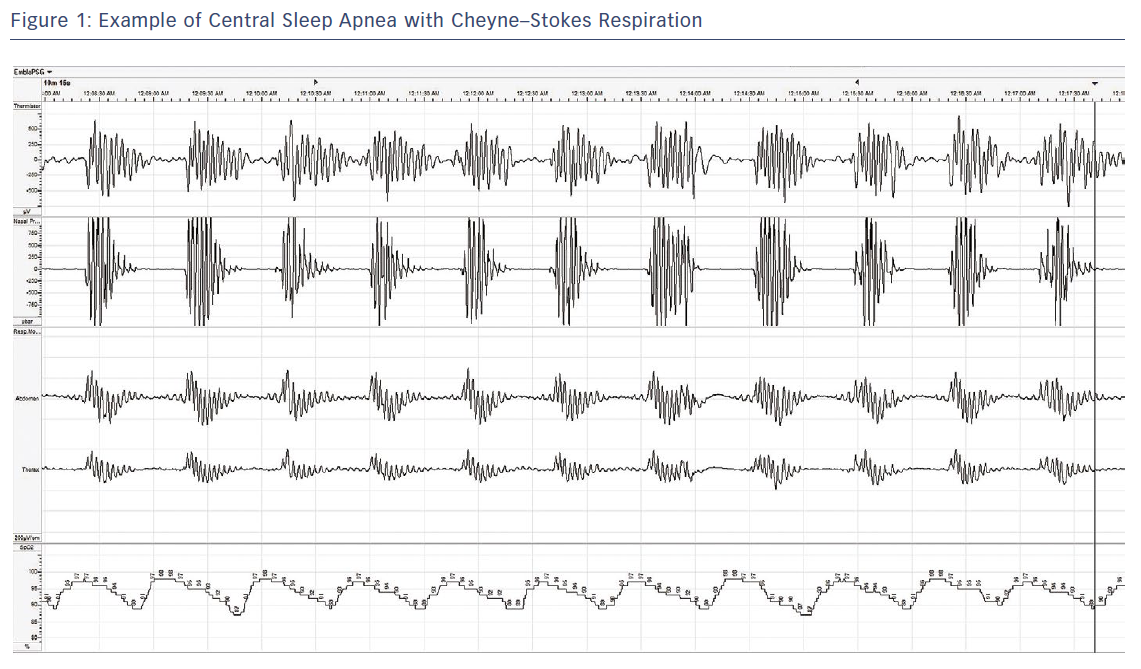
CSA occurs in two primary forms, characterised by either Cheyne–Stokes respiration or non-Cheyne–Stokes respiration.17 In HF patients, the Cheyne–Stokes respiratory pattern is much more common and has a characteristic oscillatory pattern of shallow–deep–shallow breathing (Figure 1). Cheyne–Stokes respiration results from a delay in the respiratory control centre in detecting and responding to changes in carbon dioxide levels in the blood. The full mechanism is complex and still not completely understood; however, a number of factors appear to play a role. First, in patients with a reduced ejection fraction, there can be an increase in blood circulation time, which results in a delay in the time it takes blood to reach the carotid body and peripheral chemoreceptors responsible for detecting and signalling changes in blood carbon dioxide levels. Second, sympathetic activation results in increased chemosensitivity in the carotid body, further delaying the signalling. HF patients often hyperventilate chronically, and pulmonary congestion can activate pulmonary stretch receptors to cause a relative increase in ventilation. Elevated respiration can lead to a decrease in carbon dioxide and generate a signal to stop breathing.4 Regardless of which mechanism begins the cycle, each cycle perpetuates the next, allowing the abnormal breathing pattern to continue throughout the night. Each cycle results in repetitive hypoxia and arousal events, leading to long-term detrimental effects.
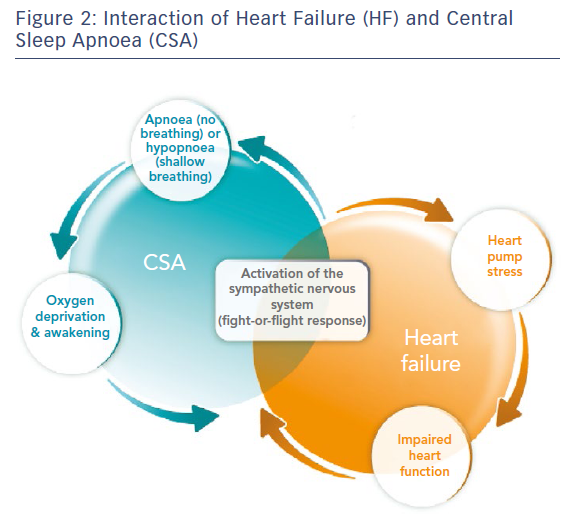
Regardless of whether the pattern is Cheyne–Stokes or non-Cheyne–Stokes, the effects of the breathing disorder result in a number of devastating effects to the cardiovascular system. Intermittent hypoxia leads to inflammation, tissue ischaemia and endothelial dysfunction. Chronically, this results in thrombosis, left ventricular (LV) hypertrophy and adverse remodelling. Each arousal results in a discrete release of norepinephrine. Norepinephrine can lead to cardiac myocyte apoptosis, cardiac arrhythmias, sodium retention and activation of the renin–angiotensin system. Long term, these effects contribute to the downward cycle of HF; therefore, it is not surprising that patients with HF and CSA have an increased risk of recurrent hospitalisation and death (Figure 2).4,12
Therapeutic Options
A number of therapeutic approaches have been utilised for the treatment of CSA. Medications such as theophylline and acetazolamide were shown to decrease the number of apnoea and hypopnoea episodes per hour in small studies, and aided significantly in demonstrating the importance of carbon dioxide in the disease process. However, neither medication has been studied in long-term, randomised controlled trials and there are risks in the HF population with these medications.4 Trials with nasal oxygen demonstrated an improvement in the number of episodes, reduced hypoxia and improved QOL, but clinical trial results have not been consistent. In addition, the risk of hyperoxia is increasingly recognised as harmful in patients with cardiovascular disease.18–20 Studies with newer oxygen delivery systems, which maintain a normal oxygen saturation without the resulting hyperoxia, may hold promise in the future. To date, the most widely used therapeutic approach, PAP, was borrowed from the treatment of OSA. Designed to open closed airways, this therapeutic approach can improve the number of events per hour, but can also worsen or unmask CSA in some patients.4,21
PAP therapies have been tested for the treatment of CSA. The largest randomised controlled trial with the use of continuous PAP (CPAP; single level of air pressure delivered throughout the night) was the Canadian CPAP for Patients with CSA and HF (CANPAP) trial.5 This study randomised 258 patients with HFrEF (ejection fraction <40 %) to CPAP therapy versus no CPAP, with a primary endpoint of morbidity and mortality. Following an interim analysis, a decision was made to stop the study due to an early increase in mortality in the treatment group and a slow enrolment. By the time the study was stopped, there was no difference between the treatment and control groups in morbidity or mortality. The study did demonstrate a reduction in AHI of 21 ± 16 events/hour (from a baseline of 40 ± 17 events/hour).5 However, a post hoc analysis showed that patients who responded to CPAP therapy (defined as achieving an AHI <15) had a decreased mortality rate compared with both CPAP non-responders and controls.22 However, no prospective studies confirming this finding have been completed to date.
Following the CANPAP trial, a new type of PAP therapy designed to improve the treatment of CSA by adjusting the pressure delivered, called adaptive servo-ventilation (ASV), was developed. It uses both inspiratory and expiratory pressure, and titrates the pressure to maintain a patent airway and adequate air movement. This therapy was studied in a large clinical trial to improve morbidity and mortality. The Treatment of Predominant CSA by Adaptive Servo Ventilation in Patients With HF (SERVE-HF) trial randomised 1325 patients to ASV plus medical treatment versus medical treatment alone.6 It was an event-driven trial and completed the full number of events pre-specified. However, the trial failed its primary endpoint, with no improvement in mortality or morbidity observed. Similarly, there was no improvement in QOL or arousals, even though AHI improved from 31.2 to 6.6 events/hour (range 0.0–71.9 events/hour). More concerning, cardiovascular mortality, a secondary endpoint, increased in the treatment group (29 versus 24 %; hazard ratio 1.34; 95 % CI [1.09–1.65]; p=0.006). A contraindication was put in place for HFrEF patients by the US Food and Drug Administration (FDA), and the American Academy of Sleep Medicine supported the contraindication in this patient population.23 Recommendations from the FDA, American Association of Sleep Medicine, and two major ASV device manufacturers suggested that patients newly diagnosed with CSA in the setting of HFrEF should not be treated with ASV device therapy. In addition, all current CSA patients with HFrEF currently on ASV therapy would need to be seen in clinic to discuss risks, options and alternatives. Further analysis demonstrated that the increase in mortality was driven by an increase in sudden cardiac death that occurred not only at night but also during the day.24
The SERVE-HF investigators suggested that this unexpected finding could be due to CSA actually being compensatory or that it could be due to the PAP therapy itself.6 CSA may begin as a compensatory mechanism similar to tachycardia; however, chronic intermittent hypoxia and increased sympathetic drive have been demonstrated to be detrimental in HF patients, and thus it is unlikely that CSA is beneficial chronically.25
Alternative theories have included a discussion of the study design itself. The design allowed crossover, and a significant number of patients did switch; 168 ASV recipients stopped therapy and 87 controls left the study, most of whom are suspected to have subsequently started ASV treatment.26 Adherence to the therapy was poor in the treatment group, with an average nightly use of 3.7 hours. However, a recently published on-treatment analysis continues to demonstrate the cardiovascular risk of the therapy.27 Also, due to the study design, cardiac implantable electronic device data were not collected, limiting adjudication of the specific cause of death. One signal from the trial was that a Cheyne–Stokes breathing pattern was associated with an increased risk of mortality, but there is no clear methodology on how this parameter was determined and no core lab scoring of these data.6
The algorithm of the specific ASV device has been hypothesised to be the cause of the increased mortality.26 One theory is that the specific ASV device utilised in the trial delivered minute ventilations at up to five-times normal in some patients, which could lead to metabolic derangements and sudden death.27 Another theory is that the pressures in some patients were much higher than expected, which could increase pressure to the right heart.28 A relationship was noted between lower ejection fraction and an increased risk in mortality.6 While right ventricular (RV) function was not analysed, it often correlates with LV dysfunction. Increased pulmonary pressure on an already weakened right heart could explain the increased mortality in the trial. Chronically increased pulmonary pressures can stress patients with right heart failure, leading to rapid and progressive RV dysfunction and sudden death.25
Following the SERVE-HF Trial, most other research with ASV was stopped. However, the Effect of ASV on Survival and Hospital Admissions in HF (ADVENT-HF) study (ClinicalTrials.gov identifier: NCT01128816), which is examining a type of ASV therapy with an algorithm that should maintain lower pressures, continues. It is enrolling both OSA and CSA patients with a reduced ejection fraction and may give some additional insight into the treatment of CSA. The data and safety monitoring board has reviewed the data several times and has decided to continue the study in both patient groups.29 Another trial, Cardiovascular Outcomes with Minute Ventilation-Targeted ASV Therapy in HF (CAT-HF), enrolled patients acutely hospitalised for HF and placed them on ASV.30 The study used the same device as in SERVE-HF and was stopped early due to the overlap in patient populations once the results of SERVE-HF were reported. However, there were some encouraging data in patients with HF and preserved ejection fraction, although the sample size was very small (n=24).
Regardless of the mechanism, PAP does not address the physiological nature of CSA: the delay in neurological response to alterations in carbon dioxide levels. The FDA recently approved a therapeutic approach to treat CSA by replacing the delayed signalling. Phrenic nerve stimulation uses neurostimulation to stimulate a single phrenic nerve at night, which restores a normal breathing pattern and stabilises carbon dioxide levels.7 The device is fully implantable, with a transvenous lead placed in a vein near one of the phrenic nerves (either the left pericardiophrenic or right brachiocephalic vein). The pulse generator is placed in either the right or left pectoral region, and respiration is monitored either via a sensing lead placed in the azygous vein or by the stimulation lead itself. The unique algorithm of the device allows it to activate automatically at night, when the patient is in a sleeping position, and suspend therapy when the patient sits up.31
A randomised controlled trial of phrenic nerve stimulation therapy versus no stimulation has been conducted.7 A total of 151 patients with CSA were implanted with the device, with 73 randomised to therapy and 78 to no stimulation. The control group had therapy activated after the primary endpoint evaluation at 6 months. The primary effectiveness endpoint was met, with a 41-percentage-point difference between groups (51 versus 11 %) in the proportion of patients achieving a ≥50 % reduction in AHI from baseline to 6 months. All prespecified and hierarchically tested secondary endpoints were met, including improvements in oxygenation (oxygen desaturation index 4 %), arousals, REM sleep and QOL measured by two different metrics: Epworth Sleepiness Scale and Patient Global Assessment.
Safety was closely monitored by independent clinical events committee and a data and safety monitoring board. The primary safety endpoint of freedom from implant-, device- or delivered-therapy-related serious adverse events was achieved in 91 % of patients. Although participants were not required to have HF, 64 % had a history of HF. Owing to the close relationship between HF and CSA, the HF subgroup was carefully studied in light of the results of SERVE-HF. Importantly, no trend of an increase in cardiovascular mortality was seen, although the trial was not powered to evaluate this outcome. Given that phrenic nerve stimulation does not employ PAP therapy to treat CSA, the possible negative effects of PAP therapy noted above should be avoided with phrenic nerve stimulation. Importantly, the FDA approved the use of phrenic nerve stimulation in moderate to severe CSA in adult patients, without regard for the presence of HF or ejection fraction, making phrenic nerve stimulation the only approved treatment in the US for CSA in HF patients with ejection fraction ≤45 %.
Conclusion
CSA clearly has chronic and detrimental effects on HF patients. While symptoms may be difficult to identify, there is clear benefit in QOL with treatment of CSA using phrenic nerve stimulation, but this has not been observed with other therapies. It is common for patients not to articulate their symptoms or attribute symptoms of CSA to their HF, which delays diagnosis and treatment of CSA. CSA results in chronic sympathetic nervous system activation and hypoxia. While it is logical that treatment of CSA will be able to decrease the significant morbidity and mortality associated with sleep apnoea, no therapy to date has demonstrated improvements in cardiovascular outcomes. However, this does not mean the disease should not be treated. Improving QOL in HF patients is extremely valuable while we wait for data demonstrating that improvements in sleep apnoea events, oxygenation and arousals lead to improvements in cardiovascular outcomes in this population.