








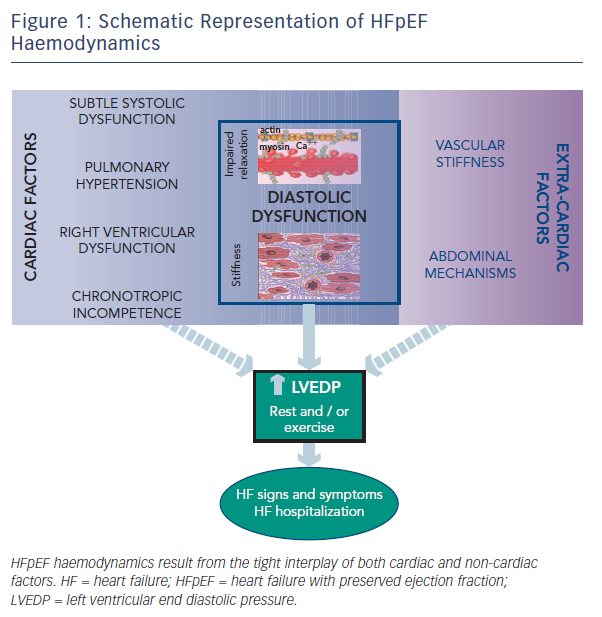
Despite the burden of heart failure (HF) with preserved ejection fraction (HFpEF),1 its pathophysiological mechanisms remain controversial and are likely to be multifactorial.2,3,4 The lack of a comprehensive paradigm applicable to all patients suggests that haemodynamic derangements responsible for this disorder may be quite heterogeneous. As recently highlighted, haemodynamic features of HFpEF involve both cardiac and extra-cardiac mechanisms. Studies on HFpEF have shown diastolic abnormalities, subtle systolic dysfunction, pulmonary hypertension, right ventricular dysfunction and chronotropic incompetence, in addition to ventricular–vascular mechanisms and abdominal factors. In this short review we highlight and discuss the different mechanisms characterizing HFpEF haemodynamics, which finally lead to elevated left ventricular end diastolic pressure (LVEDP), a common hallmark of this multifaceted syndrome (see Figure 1).
Cardiac Factors in HFpEF Haemodynamics
Patients with HFpEF are considered to be predominantly elderly women with hypertension, left atrial enlargement, obesity, and with specific pathophysiological abnormalities in cardiac structure such as myocyte hypertrophy, interstitial fibrosis, inflammation, and microvascular dysfunction. Thus, it has been postulated that the signs and symptoms of HF may be primarily a consequence of progressive abnormalities in diastolic function domains.5 The importance of diastolic left ventricular dysfunction in HFpEF has been confirmed by the majority of invasive and non-invasive haemodynamic studies, which show uniform presence at rest of slow active left ventricle (LV) relaxation and elevated passive LV stiffness.5,6,7,8 Furthermore, it has been demonstrated that elevated diastolic LV stiffness may limit cardiac performance during exercise-associated tachycardia or rapid pacing.9 In contrast, Kawaguchi et al, using the multi-beat conductance catheter technique, demonstrated in a small group of patients and controls that ventricular-arterial stiffening was the predominant mechanism underlying elevated LVEDP. However, they did not show differences in relaxation and stiffness in subjects with HFpEF compared with HF-free controls.10 Therefore, other intrinsic mechanisms have been advocated in HFpEF haemodynamics, such as subtle systolic dysfunction, pulmonary hypertension and right ventricular (RV) dysfunction.
While left ventricular ejection fraction (LVEF) is the most widely used index of systolic function, applied to distinguish HFpEF from mid-range and reduced LVEF HF patients,11 it is dependent on loading conditions and chamber size. Therefore, it is a poor measure of contractility. Importantly, it has been shown that HFpEF patients – despite the preserved LVEF – have subtle systolic dysfunction at rest, by means of reduced LV strain at echocardiographic imaging, and this dysfunction has prognostic relevance.12,13 Furthermore, it has been suggested that contractile dysfunction may contribute to inadequate myocardial response to efforts, leading to the appearance and aggravation of HF symptoms.14,15 Indeed, a recent study in HFpEF subjects examined cardiac systolic reserve during exercise and found that contractility increases were depressed.16 Therefore, the exercise test may unmask mild deficits in systolic function in HFpEF.
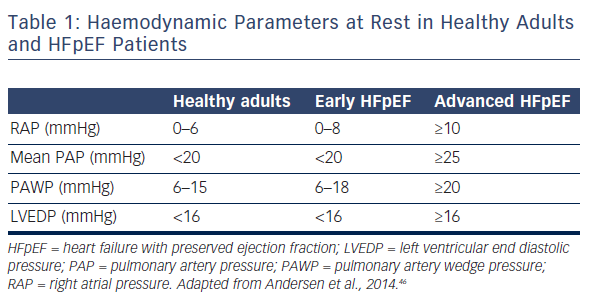
Previous studies have also reported a high prevalence of pulmonary hypertension (PH) in HFpEF.17 Importantly, PH portends worse outcome in HFpEF patients.18,19 It has been shown that the degree of PH is similar in subjects with HFpEF and HF with reduced ejection fraction (HFrEF), and is largely reversible with acute infusion of sodium nitroprusside. However, the combined potentially greater risk of hypotension or depression of stroke volume suggests that vasodilator-based approaches may not be as broadly applicable to HFpEF as they are to HFrEF.20 Another study has shown that pulmonary artery systolic pressure (PASP) rises along with pulmonary artery capillary wedge pressure (PAWP) in patients with both hypertension and HFpEF.18 However, PASP remains higher in HFpEF, even when adjusting for PAWP, suggesting a pre-capillary component to PH on top of pulmonary venous hypertension (PVH).18 Distinguishing these factors may be difficult. By definition, an elevated PAWP (i.e. >15 mmHg) characterises PVH, while pulmonary arterial hypertension (PAH) is typically associated with a normal PAWP. Of note, estimation of PAWP by non-invasive methods is suboptimal. This is because the most used echocardiographic index of elevated PAWP – E/E’ – has been shown to be only modestly correlated with supine PAWP in patients with unexplained dyspnoea and preserved LVEF and, in general, this index lacks sensitivity.21,22,23 Therefore invasive evaluation currently provides more reliable diagnostic information (see Table 1). Of note, PAWP obtained at the time of right heart catheterisation is influenced by resting conditions and the patient’s volume status at the time of the procedure. Indeed, it has been demonstrated that resting haemodynamics may be normal, while performing right heart catheterisation during supine exercise may unmask a diagnosis of HFpEF.24 Furthermore, invasive exercise testing may substantially improve prediction of long-term mortality in patients with suspected HFpEF.25 Because this technique is mainly used in research institutions, a possible alternative is to perform a fluid challenge at the time of catheterisation, to confirm HFpEF presence,26 and eventually to differentiate PAH from PVH. In fact, a recent study showed that among 207 patients with a suspected diagnosis of PAH, one fifth developed elevated PAWP after a fluid bolus, and were reclassified as having PVH.27 Nonetheless, it is important to remember that approximately 20 % of normal adults may develop PAWP >15 mmHg with acute saline infusion.28 Overall, such data imply that many patients with PH may have an under-recognised component of PVH linked to left-sided HF, which is manifested more under conditions of exertion or volume loading.29 In particular, exercise stress testing or volume challenge are indicated at the time of invasive procedure for patients presenting with PH and normal PAWP who are obese and/or who have a dilated left atrium.30 However, there is not presently enough evidence on standardisation of these procedures. There are no validated cut-off values for a pathological haemodynamic response during exercise or after acute saline infusion that would allow a clear classification of HFpEF. Generating these values by performing diagnostic trials is of major importance, but at present it is difficult to recommend routine exercise haemodynamic testing and/or a volume challenge for clinical practice.
Another invasive haemodynamic study has recently shown that RV dysfunction is common in HFpEF and is caused by both RV contractile impairment and afterload mismatch from PH.31 In this study, the factors associated with RV dysfunction were increasing pulmonary artery pressures, atrial fibrillation, male sex and LV dysfunction. It has also been demonstrated that patients with HFpEF display impaired RV reserve during exercise that is associated with high filling pressures and inadequate cardiac output responses.32 These findings highlight the importance of biventricular dysfunction in HFpEF haemodynamics.
Chronotropic incompetence represents another important characteristic of HFpEF, which has been described in approximately 30 % of patients.33,34,35 Indeed, chronotropic reserve is depressed in HFpEF even when compared with older, age-matched controls, independently from rate-lowering medication use. Chronotropic incompetence may help to partially explain why most patients with HFpEF complain of symptoms only during physical exertion. Since the increase in plasma catecholamine with exercise is similar in HFpEF and healthy controls, it has been suggested that chronotropic incompetence may be related to deficits in beta-adrenergic stimulation.33 Additionally, autonomic dysfunction may be a contributing factor, as heart rate recovery is abnormal and baroreflex sensitivity impaired in HFpEF.34
Extrinsic Factors in HFpEF Haemodynamics
An alternative model for HF development in HFpEF patients underscores the contribution of non-myocardial factors to systolic and diastolic LV performance.
Cardiac function is affected by the net balance between afterload and preload.36 Central aortic stiffness, increasing systolic load and negatively affecting ventricular–vascular coupling, may accelerate HF development in at-risk patients. Of note, aortic stiffness increases with age, particularly in women with hypertension, and is a precursor of incident HF.37,38 To preserve adequate coupling among the heart and the arterial system, ventricular systolic stiffening also increases, and this combined ventricular–vascular stiffening is a hallmark of HFpEF.10,39 This limits LV systolic reserve, increases the cardiac energy demands required to enhance cardiac output, and plays a central role in arterial pressure liability accompanying small changes in LV preload.10
Other extrinsic factors advocated in HFpEF haemodynamics comprise abdominal mechanisms. In many HFpEF patients fluid retention may occur in the abdominal cavity, with bowel congestion leading to endotoxin translocation and systemic inflammation. In the same way, systemic inflammation may also be induced by comorbidities such as obesity, diabetes mellitus or chronic obstructive pulmonary disease that are highly prevalent in these patients, and this has been suggested as a possible cause of myocardial structural and functional alterations.40,41 Of note, the increased neurohormonal activation typical of HF results in venoconstriction, with impaired capacitance function of the splanchnic vasculature. Increased capillary hydrostatic pressure leads to a rise in intra-abdominal pressure and eventually also to organ dysfunction. Indeed, HFpEF is frequently associated with renal impairment, as chronic kidney disease (CKD) occurs in up to two thirds of HFpEF patients and is associated with poor prognosis.42,43,44 There is a bidirectional link between HFpEF and CKD, as it has been shown that venous congestion may lead to CKD and, vice versa, renal impairment begets congestion and HF. Renal impairment causes metabolic derangements and affects circulating factors causing an activated systemic inflammatory state and endothelial dysfunction. This may lead to hypertrophy, myocardial stiffening, and interstitial fibrosis via cross-talk between the endothelium and cardiomyocyte, finally causing haemodynamic impairments.45
HFpEF Haemodynamics: Critical Appraisal
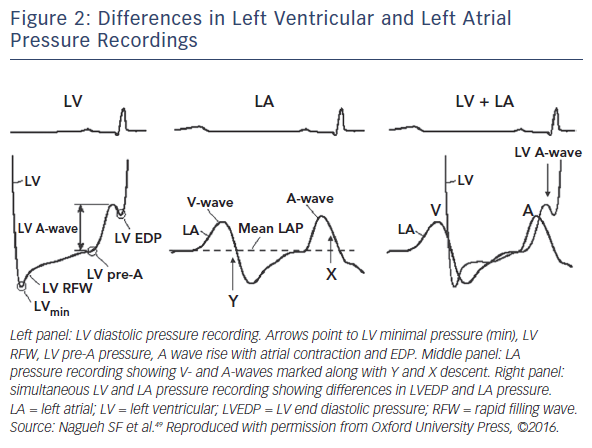
While diastolic dysfunction was originally considered pathognomonic in HFpEF haemodynamics,5,6,7,8,9 abnormalities of active LV relaxation and/or passive stiffness have been described by most but not all invasive haemodynamic studies.10 There are different reasons for these discrepancies, such as heterogeneity in study populations, differences in control groups, the use of invasive or non-invasive studies, or examinations being performed only at rest or also during effort. Additionally, a major challenge to the field is that truly representative experimental models of HFpEF do not exist, and human data – particularly direct myocardial analysis – remain limited, with very small populations having been studied. There are no data from beating muscle or cells from human hearts, and existing animal models fail to capture the complexity of the human disease. Furthermore, it should be emphasised that only left heart catheterisation allows for direct measurement of LVEDP, as well as the kinetics of relaxation and passive chamber stiffness through pressure-volume recordings (PV loops). However, these assessments require highfidelity micromanometer and conductance catheter systems, which are demanding techniques that are not easily reproducible and not widely available in clinical settings. Of note, even though it has been suggested that a high-quality PAWP tracing is just as robust as directly measured LVEDP,46 this assumption is incorrect in the case of LV disease. This is because a strong atrial contribution to LV filling can occur in this condition, which can translate in a LVEDP considerably higher than the mean left atrial pressure and PAWP (see Figure 2).47 Overall, these considerations seem to suggest that diastolic dysfunction may still have a central role in HFpEF haemodynamics, although it may be demanding to prove it in clinical and experimental studies (see Figure 1).
Finally, the recognition of diastolic LV dysfunction as the predominant mechanism underlying HFpEF haemodynamics does not necessarily imply that the latter represents the sole contributor to haemodynamic derangements. Indeed, numerous other mechanisms have been recently identified and may play important roles. Among these, both cardiac and extra-cardiac factors should be assessed to adequately interpret HFpEF haemodynamics. However, it should be emphasised that ultimately most of these mechanisms negatively affect LVEDP, which represents the hallmark of HFpEF, together with normal LV dimensions and preserved LVEF. Raised LVEDP justifies HF signs and symptoms, may lead to myocardial ischemia, fibrosis, and structural impairment, and portends adverse prognosis in HFpEF patients. An approach centred on the specific pathophysiological abnormalities in cardiac structure and function, in particular diastolic dysfunction, may be more effective for therapeutic discovery.48