










Heart failure with preserved ejection fraction (HFpEF) is becoming the most common cause of heart failure worldwide, in part, driven by a rising prevalence of obesity.1 Since the first description of obesity-related HFpEF, there has been widespread recognition of this being a unique phenotype with its pathophysiology driven directly by excess adiposity.2–4 Notably, obesity is the predominant driver of an increasing prevalence of HFpEF in younger people.5,6 Although generalised and visceral adiposity is important in the pathogenesis of obesity-related HFpEF, there is increasing recognition of the potential role of epicardial adipose tissue (EAT) in disease pathogenesis. EAT is metabolically active tissue located directly on the surface of the myocardium underneath the visceral pericardium. By virtue of its anatomical interface with the heart and the lack of fascial separation between the underlying myocardium and epicardial fat, locally secreted adipokines directly bathe the surface of the heart and result in underlying myocardial remodelling.7 Its position on the surface of the myocardium allows EAT to directly contribute to an increase in total heart size with stretch of the pericardium and results in relative pericardial restraint with constrictive physiology.3,8,9 EAT is most commonly measured by echocardiography in the parasternal long axis view perpendicular to the right ventricle (RV) to quantify epicardial fat thickness and this has been correlated with worse haemodynamic derangements and adverse outcomes in HFpEF.9,10 Alternatively, cardiac MRI or CT can provide a more complete volumetric assessment of epicardial fat volume and has also demonstrated associations with adverse outcomes and functional metrics in most but not all HFpEF studies (Table 1).11–15
Epicardial fat has been independently associated with abnormal cardiac structural changes and incidence of HFpEF in community-based studies further supporting its potential causal role.16,17 Notably, these studies have demonstrated independent associations between baseline epicardial fat with risk of heart failure in both men and women, which persisted even after adjustment for various anthropometric measures, including overall BMI, visceral fat and biomarkers of inflammation.17 There is, however, a strong collinear relationship between generalised obesity and epicardial fat, making the relative contributions of epicardial fat compared to overall adiposity in the pathogenesis of HFpEF less clear even after statistical adjustment.11,12 Therefore, it remains unclear if EAT truly independently contributes to obesity-related HFpEF or is just a marker of severe obesity. In this review, we will discuss the potential pathophysiology of epicardial fat as it relates to HFpEF, potential therapies targeting EAT with implications for HFpEF and the arguments for and against its causal role.
Does Epicardial Adipose Tissue Have a Causal Contribution to Obesity-related HFpEF?
Epicardial, visceral and overall adiposity are clearly linked with incident HFpEF but also with AF and atrial myopathy, which are important components of the HFpEF syndrome.18–23 Recent studies focusing on EAT have elucidated some of its key roles in obesity-related HFpEF. EAT can theoretically be a contributor to obesity-related HFpEF by either mechanically enhancing pericardial restraint and/or through local secretory effects from its direct contact with the underlying myocardium causing adjacent ventricular and atrial remodelling.2,9,24–26
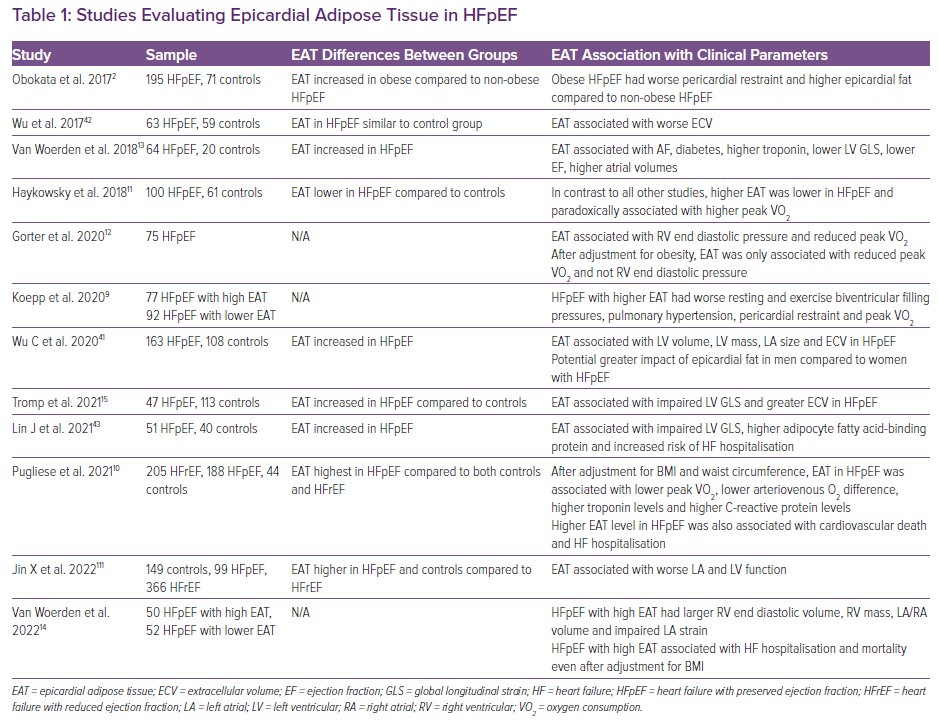
The pericardium normally exerts a compressive contact force on the heart that helps couple left and right ventricular preload to try and maintain a constant stroke volume in the face of changing venous return. As with any chamber in the heart, the pericardium has its own curvilinear pressure–volume relationship with an initial flat portion followed by an exponential increase when stretched to the steep portion of its pressure-volume relationship.8,27 In patients with substantial obesity with cardiomegaly and increased epicardial fat volume, the pericardial sac is stretched to a steeper pressure-volume relationship and there is enhanced diastolic ventricular interaction and pericardial restraint (Figures 1 and 2). This exerts an exaggerated compressive force on the heart resulting in higher left- and right-sided filling pressures for any degree of left ventricular (LV) end diastolic volume – the true LV preload.
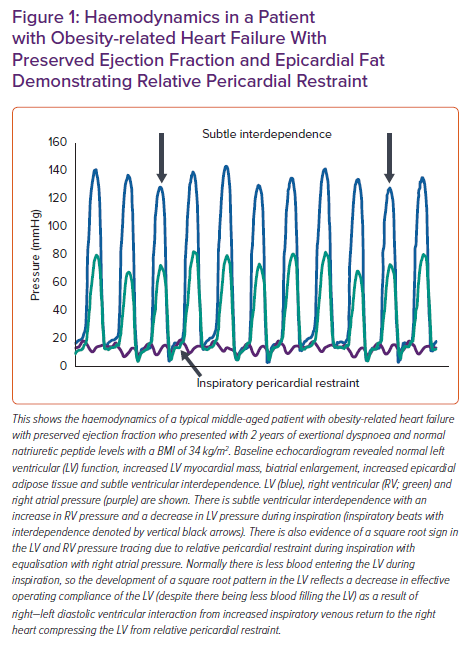
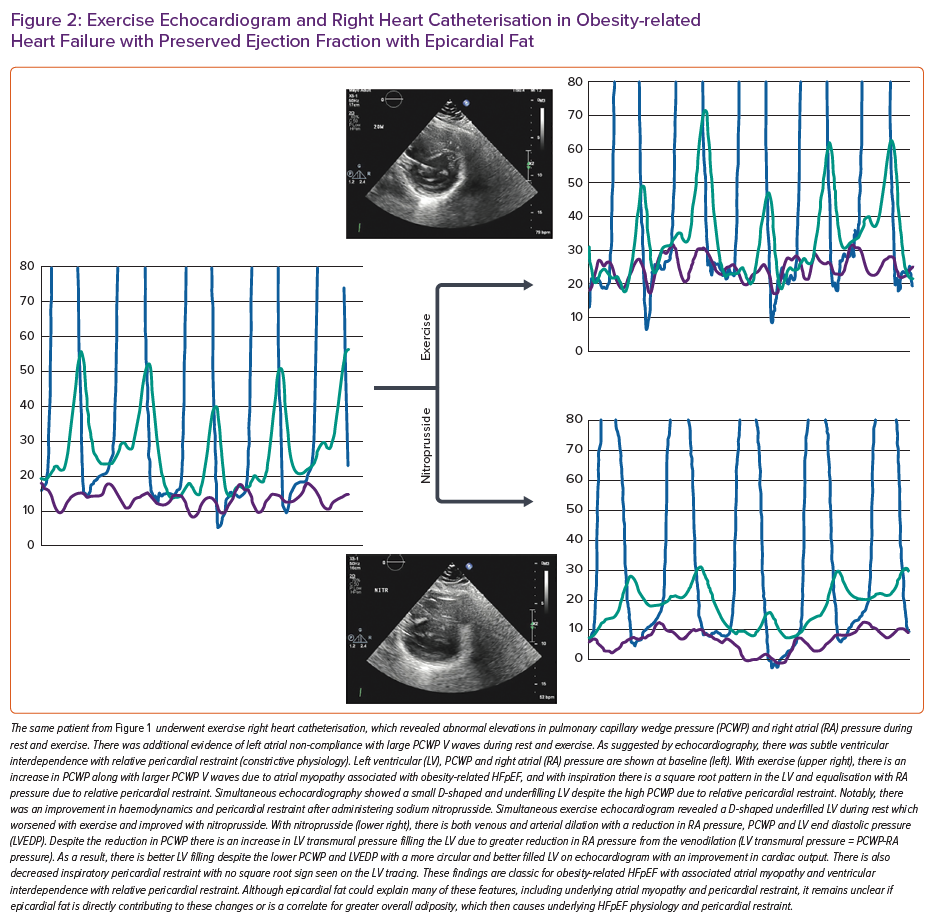
Although gradual cardiac enlargement can allow some degree of pericardial remodelling and improved pericardial compliance, eventually such reserve is exhausted, particularly during exercise when there is a rapid bolus of venous return to an already overstretched pericardial sac.28,2 Since EAT lies between the visceral pericardium and underlying myocardium, its expansion can result in a stretched pericardium with resultant compressive contact force on the heart, independent of the underlying heart size and intrinsic pericardial properties. This compressive force increases pulmonary capillary wedge pressure (PCWP) for any LV end diastolic volume and is associated with higher right atrial (RA) pressure (which closely approximates pericardial pressure in man) (Figure 3).29 Functionally, this results in a heightened RA/PCWP ratio during rest and exercise and an increased eccentricity index on echocardiography (D shaped LV), which compromises LV filling and cardiac output response during exercise despite a pathologically elevated PCWP.8 This is further reinforced by the finding that filling pressures in obesity-related HFpEF are higher in patients with HFpEF and greater EAT.9 Even if the PCWP does rise primarily due to constrictive physiology, this still contributes to pulmonary congestion and dyspnoea during exercise, and in fact patients with higher RA pressures, such as those with pericardial restraint, have the greatest pulmonary congestion during exertion resulting from impeded lung lymphatic drainage into a systemic venous system with high venous pressure.30–33 Multiple other studies have also correlated epicardial fat with peak oxygen consumption (VO2) and haemodynamic derangements in HFpEF consistent with a potential contributory role (Table 1).9,13,15
Epicardial fat has also been associated with AF even in HFpEF patients, with biological plausibility for a causal relationship through underlying myocardial injury, inflammation, and adjacent atrial and ventricular remodelling.13,23,25,26,34–39 This atrial and ventricular remodelling may relate to direct secretion of adipokines and inflammatory profibrotic markers from the epicardial fat, thereby affecting the underlying myocardium through activin-A, angiopoietin-2, interleukins, tumour necrosis factor α and infiltration of inflammatory cells such as monocytes and macrophages.24,26,35,36 Meta-analyses have demonstrated an association between EAT and diastolic dysfunction.40 A number of HFpEF studies have also demonstrated a correlation between EAT with worse exercise capacity, myocardial injury, impaired myocardial structure and function, extracellular volume suggestive of fibrosis and impaired resting and exercise haemodynamics (Table 1).9,13–15,41,42
A study that specifically looked at exercise haemodynamics in HFpEF patients with and without increased EAT demonstrated worse resting and exercise biventricular filling pressures, pulmonary hypertension and pericardial restraint along with impaired exercise capacity.9 Longitudinal studies have demonstrated an independent relationship between EAT and incident heart failure, even after adjusting for overall BMI, fat distribution and sex suggesting a causal relationship.17 Similarly, EAT in overt HFpEF patients appears to be associated with hospitalisation for heart failure and mortality risk, even after adjusting for BMI.10,14,43 EAT in community participants is also associated with preclinical abnormalities in atrial and RV strain and a worse 6-minute walk distance which further suggests the potential to predispose to early HFpEF.16 Additionally, epicardial fat appears to be associated with atherosclerosis progression and is linked with incident HFpEF even in patients with coronary disease suggesting that coronary endothelial dysfunction and atherosclerotic changes may also mediate some of the links between HFpEF and epicardial fat.44,45 Thus, there is a body of evidence supporting a potential independent role for epicardial fat in the pathogenesis of HFpEF and specific therapies directed towards EAT require further investigation to both prevent and treat HFpEF.
Is EAT Merely a Marker of Functionally Severe Obesity in HFpEF?
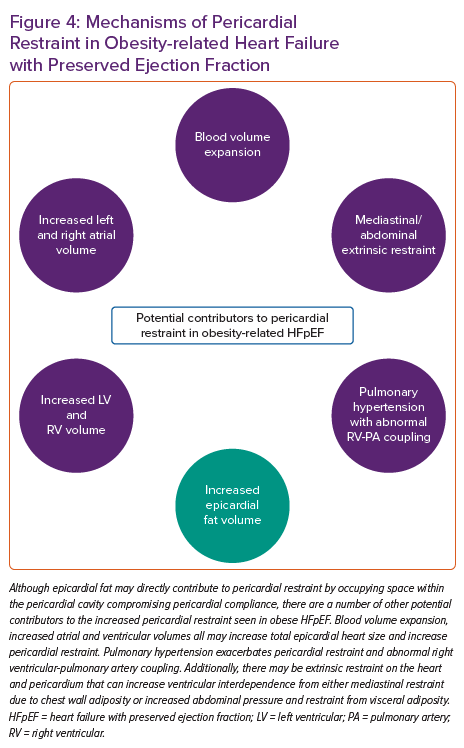
Although EAT appears to be associated with abnormal haemodynamics that are pathognomonic of HFpEF, the presence of EAT is also associated with diabetes and greater visceral and overall adiposity, which have also been associated with HFpEF and this makes a direct determination of causality with HFpEF difficult.9,13,14,18,20,46,47 Therefore, while there is a growing body of literature to suggest a strong association between EAT and obese HFpEF, there remains difficulty in establishing causality given the strong collinear associations between epicardial fat, visceral fat and total body fat.11,12 In other words, it remains unclear if the presence of EAT is merely a correlate of the generalised adiposity-related inflammation that drives the obese HFpEF syndrome, or if excess EAT directly contributes to HFpEF pathogenesis independent of visceral and systemic adiposity.3,48,49 Patients with increased EAT and HFpEF generally are more obese with larger blood volumes and greater potential for generalised obesity-related HFpEF haemodynamic derangements.2,9,12 Notably, although epicardial fat appears to be associated with worse pericardial restraint in HFpEF, there are multiple other reasons for increased pericardial restraint in obese HFpEF, including global cardiomegaly, mediastinal restraint, increased blood volume, abdominal compression from visceral adipose tissue and pulmonary hypertension with abnormal RV-pulmonary artery coupling (Figure 4).9 High filling pressures in obesity-related HFpEF are proportional to body mass and associated plasma volume, all of which distend the heart and pericardium exacerbating relative pericardial restraint.2,50
EAT is also clearly associated with visceral adipose tissue and tends to track visceral adipose changes, as seen with bariatric surgery where substantial weight loss results in regression of both fat depots.51 Furthermore, many patients still develop HFpEF despite having normal amounts of EAT. Therefore the presence of EAT is not essential for the manifestation of exertional pulmonary venous hypertension that is pathognomonic of HFpEF. 9,52,53 In addition, the only randomised trial of weight loss in HFpEF demonstrated that symptoms and exercise capacity could be improved with modest weight loss in obese HFpEF and this clinical improvement was associated with a regression in visceral and subcutaneous adipose tissue but with no measurable change in epicardial fat by MRI.54 Large weight loss results in improved haemodynamics in obese patients, which may be only partially related to epicardial fat regression, but, regardless of the mechanism, weight loss through bariatric surgery clearly prevents incident heart failure that is likely to be HFpEF.51,55,56 Therefore, epicardial fat does not appear essential for HFpEF in all patients and obese HFpEF patients still experience clinical improvement with weight loss in the absence of epicardial fat modulation.2,9 Although there may be subsets of obese HFpEF patients where epicardial fat is directly causal in part of their pathophysiology, it remains difficult to conclusively delineate the independent causal contribution of EAT to obesity-related HFpEF. Regardless of causality, the mere presence of epicardial fat in a patient with HFpEF is a biomarker associated with worse exercise haemodynamics, pericardial restraint, exercise tolerance and prognosis and is an important clinical feature to identify.9,14
Therapies Targeting Epicardial Fat
Lifestyle Modification
Substantial weight loss, such as that associated with bariatric surgery, has been associated with reduced epicardial fat thickness along with regression of visceral and generalised adiposity.51,57 In contrast to this data with bariatric surgery, modest weight loss just through diet change and exercise in obese HFpEF in the SECRET trial did not decrease epicardial fat volume.54 Apart from general recommendations for patients to lose weight (which are generally ineffective), the CENTRAL randomised trial demonstrated that a Mediterranean-style diet high in unsaturated fats with low carbohydrate intake decreased epicardial fat, compared to a low-fat diet, independent of absolute weight loss achieved.58 However, physical activity alone failed to achieve epicardial fat reduction similar to findings from the SECRET trial.54 Since a Mediterranean-style diet has also been shown to reduce adverse cardiovascular outcomes in the large PREDIMED randomised trial, this may represent an attractive dietary intervention, specifically in obese HFpEF patients, for cardiovascular risk reduction and for its direct effects on epicardial fat and central haemodynamics.59 In the CENTRAL trial, although physical activity by itself failed to affect epicardial fat, it did appear to enhance the effect of the Mediterranean and low-carbohydrate diet on epicardial fat loss, in addition to contributing to loss of visceral adiposity with all the associated metabolic benefits, and therefore the combination may be the most efficacious.58
The SECRET trial in obese HFpEF, as discussed above, used a low-calorie diet without the Mediterranean component and this failed to affect epicardial fat, which suggests that it may be the Mediterranean diet and increased unsaturated fats that specifically modulate epicardial fat reduction.54 Even without epicardial fat reduction, a low-calorie diet in obese HFpEF was associated with reduced overall adipose mass, improved quality of life, peak VO2, 6-minute walk distance, decreased serum inflammatory markers and reduced LV mass, and an increased E/A ratio suggestive of improved diastolic filling, with less impressive results seen with exercise training only.54 The combination of diet and exercise again appeared to be additive. These results contrast previous prospective observational research demonstrating reductions in epicardial fat with a low-calorie diet where more weight loss was achieved.60 Notably, weight loss was modest in the SECRET trial and generally <10 kg, which is much lower than that achieved with bariatric surgery. Additional research is needed to understand the relative benefit of specific diet interventions in obesity-related HFpEF, the relationship between the complex reduction in epicardial fat (as opposed to visceral and subcutaneous fat deposits) and epicardial heart size, and how this relates to an improvement in symptoms, exercise capacity and pericardial restraint.
Given the emerging role of obesity and potentially epicardial fat in the pathogenesis of AF, the observation of dramatic decreases in AF recurrence with weight loss are of great interest in obesity-related HFpEF. 13,23,26,34,38,39,61–64 Although these studies did not specifically include patients with HFpEF, the diagnosis of HFpEF in the presence of symptomatic AF is particularly challenging in the presence of obesity.2,52,65 It is likely that many patients in these studies had underlying occult HFpEF, which would have been confirmed with exercise testing, particularly since HFpEF is extremely common in symptomatic patients with obesity and AF.52,53,65–69 How much of the reported weight loss benefit on AF recurrence is related to improvements in haemodynamics and the local inflammatory secretory profile from loss of epicardial fat remains to be determined. Permanent AF is also liable to further worsen relative pericardial restraint from progressive severe biatrial enlargement based on animal and human studies providing further rationale for prevention of AF progression through regression in epicardial fat, but direct confirmation of these results is lacking.70–72
Bariatric Surgery
Bariatric surgery is the most effective weight loss therapy currently available but also has independent benefits on epicardial fat deposits, which may have advantages in terms of relieving pericardial restraint.51,57 Independent of these changes in epicardial fat, there are also reductions in blood volume, LV mass, cardiac output and LA size, all of which would be expected to further improve relative pericardial restraint through a reduction in total epicardial heart size.73 These reductions in blood volume and heart size are associated with reduced biventricular filling pressures suggestive of the alleviation of pericardial restraint.55 The reduction in epicardial fat deposits may also provide the independent benefit of reducing proinflammatory and profibrotic signalling pathways affecting the underlying adjacent myocardium.24,26,47 Since epicardial fat is associated with the future risk of HFpEF and heart failure hospitalisation, it is likely that some of the observed benefits of bariatric surgery on improving haemodynamics and decreasing incident heart failure are mediated by the observed reduction in epicardial fat.14,17,51,55–57
Drug Therapies Targeting Epicardial Fat and Implications for HFpEF
Very little is understood about the impact of medical modulation of epicardial fat in HFpEF. The first proven agents to improve heart failure hospitalisation and quality of life in HFpEF are the sodium–glucose cotransporter-2 inhibitors (SGLT2i).74–76 Although the mechanisms of benefit of these drugs are uncertain, they have demonstrated a reduction in epicardial fat despite only minimal weight loss suggesting a direct lipolytic effect on epicardial fat.77–79 The use of SGLT2i has also been associated with reduced incident AF, which may, in part, be due to the reduction in epicardial fat.80,81 The diuretic effect of SGLT2i may facilitate a reduction in plasma volume and mechanistic studies have shown that they also promote ventricular mass regression, which may cumulatively decrease pericardial restraint.77,82,83 Likely, in part, through these mechanisms, SGLT2i have demonstrated haemodynamic benefits on PCWP at rest and exercise in systolic heart failure and HFpEF (NCT04730947).
Thiazolidinediones, glucagon-like peptide 1 receptor (GLP-1) agonists, dipeptidyl peptidase-4 (DPP-4) inhibitors and statins have also shown efficacy in targeting epicardial fat. The thiazolidinediones, such as pioglitazone, have an effect on epicardial fat, but given their propensity to cause fluid retention, their use is contraindicated in patients with HFpEF.84 GLP-1 agonists improve cardiovascular outcomes in people with diabetes with a normal EF who are at risk of HFpEF.85–87 Liraglutide, a GLP-1 agonist, failed to improve outcomes in systolic heart failure with a possible signal of harm in heart failure hospitalisation with consistent signals towards increasing heart rates, which may explain the detriment in systolic heart failure.88–90 However, the pathophysiology of HFpEF differs profoundly from systolic heart failure and these drugs have proven benefits for cardiovascular outcomes and excellent safety data in patients with a normal EF.85–87 With the known presence of GLP receptors on epicardial fat, the effect of GLP-1 agonists on reducing epicardial fat and the potential contributory role of epicardial fat in HFpEF pathogenesis, targeting obese HFpEF patients with GLP-1 agonists may represent a high yield investigational target and provide insight into the role of epicardial fat in HFpEF.91–94 There has been recent data that higher doses than are used for diabetes of the GLP-1 agonists liraglutide and particularly semaglutide, and the novel dual glucose-dependent insulinotropic polypeptide and GLP-1 agonist tirzepatide induce profound weight loss with improved quality of life even in people without diabetes, and these drugs are being actively investigated in HFpEF (NCT05371496). 95–97
There is controversy about the safety of DPP-4 inhibitors in heart failure (leading to a Food and Drug Administration warning for saxagliptin and alogliptin) though this risk is not uniform across the drug class.89,98 Sitagliptin has demonstrated cardiovascular safety with no increased risk for heart failure and experimental evidence suggests its use can reduce epicardial fat.99–101 Therefore, further investigation of sitagliptin in obese HFpEF and its effect on epicardial fat may be warranted. Finally, statins have prominent effects on reducing epicardial fat and given their safety and proven cardiovascular efficacy in patients at risk for adverse cardiovascular outcomes and observational data supporting benefit in HFpEF, these drugs may also have a role in modulating epicardial fat in obese HFpEF.84,102–108 Pragmatically, a randomised trial testing statins in obese HFpEF would be difficult to perform as most of these patients independently meet criteria for statin therapy, especially with the outcome benefit of statin therapy in the HOPE-3 trial for which most middle-aged obesity-related HFpEF patients would meet entry criteria.106
Conclusion
Although there is a clearly demonstratable association between epicardial fat and HFpEF, the association remains highly confounded by comorbid conditions and the greater generalised obesity seen in patients with more epicardial fat. Ongoing studies testing different methods of weight loss, SGLT2i and GLP-1 agonists will provide novel insight into the potential contributory role and effect of therapies on epicardial fat in HFpEF. Further study of how epicardial fat affects cardiovascular haemodynamics and pericardial restraint in obesity-related heart failure is urgently needed given the increasing prevalence of obesity in the western world, particularly among patients with heart failure.1,2,18,109,110 For now, what is clear is that regardless of causality, the mere presence of epicardial fat is a useful biomarker for the presence of worse haemodynamics, pericardial restraint, exercise tolerance, myocardial remodelling and heart failure risk in patients with or at risk for HFpEF.