









From Acute Heart Failure Towards Worsening or De Novo Heart Failure
The natural history of heart failure (HF) is characterised by disease progression and episodes of worsening HF and acute decompensation requiring outpatient treatment intensification, emergency department or in-hospital care.
Acute HF (AHF), also known as acute decompensated HF, is defined as a progressive and sometimes rapid onset or worsening of symptoms and/or signs of HF.1 AHF may present as new onset HF (de novo HF) or worsening chronic HF (WHF), where WHF may be defined as worsening signs and symptoms requiring additional therapy. WHF represent 80–90% of HF hospitalisations.2 Compared with WHF, de novo HF patients have a different clinical profile. Generally, the patients are younger, with less previous MI and less global comorbidity burden.3 Accordingly, mortality rates are lower and the potential for improvement and possibly recovery is greater in de novo than in chronic HF (CHF).4,5 However, hospitalisation for de novo HF is still considered a critical event in the trajectory of the disease, given that mortality rates are tripled compared with patients who are never hospitalised.6
AHF is increasingly recognised as an event rather than a distinct syndrome, and this event is heterogeneous with variable onset and presentation, may increasingly be managed in outpatient day clinics or emergency departments, and may be more appropriately termed WHF. In patients with WHF, the profile of haemodynamic congestion is similar regardless of reduced (HFrEF) or preserved ejection fraction (HFpEF), but patients with HFpEF as compared with HFrEF appear to have more interstitial than intravascular fluid overload, possibly due to reduced venous capacitance and lower arterial compliance.7–9
Patients hospitalised for HF are at high risk for adverse outcomes in hospital, but also after discharge.10 In the European Heart Failure Long-term Registry (ESC-HF-LT) 1-year mortality in AHF and in chronic stable HF was 23.6% and 6.4%, respectively. Rates of death or hospitalisation for HF were 36% in AHF patients and 14.5% in CHF patients.11
Despite intensive research, no treatment has yet been shown to reduce mortality or risk of rehospitalisation in AHF.12 However, with optimal therapy it has been suggested that early rehospitalisation may be preventable in up to 70% of cases.13
Congestion
Regardless of HF aetiology, HF patients can be divided into four different profiles depending on clinical status. Patients may be described as either wet or dry, depending on their congestion status, and as warm or cold, depending on their perfusion status, with the combination of wet and cold (congested and hypoperfused) having the worst prognosis.14 Clinical signs of hypoperfusion include cold, sweaty extremities, narrow pulse pressure, dizziness, oliguria and mental confusion. Typical clinical signs of congestion include increased jugularis venous pressure, orthopnoea, pulmonary rales, peripheral oedema, third heart sound and hepatomegaly.
The terms congestion and fluid overload are often used interchangeably, however haemodynamic congestion reflects increased cardiac filling pressures, but does not necessarily equal volume overload in the extracellular compartments, particularly in the acute setting. Over time, if haemodynamic congestion continues to progress, clinical signs of congestion may evolve. In contrast, in acute pulmonary oedema, pulmonary congestion is predominantly due to an acute increase in afterload with a relative volume redistribution rather than an absolute fluid accumulation.15 Overall, congestion and fluid overload (the wet haemodynamic profile) is the most common profile in patients presenting with AHF. Less than 10% of patients present with signs of hypoperfusion and low blood pressure (the cold haemodynamic profile).16,17 Even if patients have some degree of pulmonary congestion, relatively few present with fulminant pulmonary oedema18,19 and the majority of patients instead have gradual onset of backward failure, fluid retention, congestion and often (but not always) weight gain.20–22
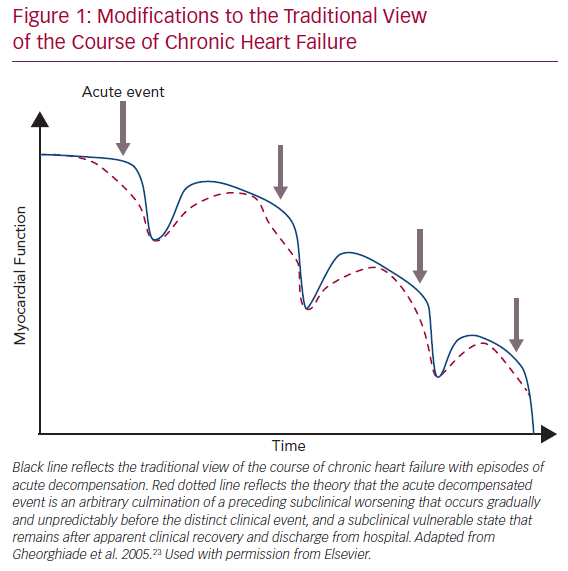
The traditional view of the course of CHF is shown by the blue line in Figure 1.23 Due to chronic maladaptive neurohormonal activation, HF progresses gradually and then some inciting event, such as an infection or poor adherence to medical treatment, causes sudden AHF needing hospitalisation. However, accumulating data from implantable devices are suggesting that the AHF event is really an arbitrary culmination of a chronic WHF that has occurred over weeks (Figure 1; red dotted line).20,24–26 Thus, AHF may be more appropriately considered WHF resulting from progressive insidious congestion. Implantable haemodynamic monitoring may be effective by recognising worsening congestion in the subclinical state, allowing prompt adjustment of therapy and averting hospitalisation.24
In the absence of mortality-reducing therapy, the main goal in WHF treatment is symptom and congestion relief. A cornerstone in the treatment of WHF and excessive volume overload is IV loop-diuretics, a therapy used in approximately 90% of AHF hospitalisations.27,28
Analogously to the insidious progressive congestion preceding WHF events, residual congestion at discharge is underrecognised and/or undertreated, exceedingly common (in one study half of patients were discharged with weight gain), and strongly associated with higher mortality and higher rehospitalisation rates.23,29–32 The traditional view holds that after WHF, the patient recovers to a point that is lower than before WHF but still represents a distinct recovery (Figure 1; blue line). We suggest that the reality is closer to hospital discharge being an arbitrary event determined only in part by clinical appropriateness, with a highly variable post-hospital course (Figure 1; red dotted line). The heterogeneity and poor treatment of WHF may be one reason recent WHF trials such as the Relaxin in Acute Heart Failure 2 (RELAX-AHF-2) trial and the Trial of Ularitide Efficacy and Safety in Acute Heart Failure (TRUE-AHF) have failed to demonstrate a clinical benefit of vasoactive and decongestive therapy.33,34 Why does congestion continue to present such a considerable clinical problem?
Euvolaemia
First, determination of euvolaemia can be challenging, given that even patients with limited signs and symptoms of fluid overload may have substantial subclinical congestion.35 Furthermore, dyspnoea relief and the patient’s return to normal body weight after treatment have been shown to be poor predictors of successful decongestion.36,37 Hence, simple clinical tools to determine euvolaemia are lacking.
Lung ultrasound is increasingly being recognised as a tool to assess pulmonary congestion and has been shown to be superior to X-ray in ruling out interstitial oedema or pleural effusions.38,39 A multiparameter-based pre-discharge evaluation of euvolaemia/residual congestion is suggested by the Heart Failure Association (HFA) of the European Society of Cardiology (ESC).10 This comprehensive evaluation including assessment of jugular venous pressure, hepatomegaly, oedema, 6-minute walk test, natriuretic peptides, chest X-ray, vena cava imaging and lung ultrasound, may be a useful tool in recognising residual congestion, but this method has not been evaluated prospectively.
Diuretic Resistance
Second, diuretic resistance, a phenomenon often seen in CHF, may hamper the success of decongestive therapy. Diuretic resistance is most commonly defined as the inability to achieve an adequate natriuretic and diuretic response despite high doses of diuretics;27,40 however, no universal definition exists. Diuretic response is a measure of decongestive effect in relation to diuretic dose, often defined as weight change per 40 mg furosemide.41 The pathophysiology of diuretic resistance is multifactorial, including the influence of neurohormonal activation, inflammation and fluctuating renal function.27 The pharmacokinetics and pharmacodynamics of the drug given is of importance for the diuretic response. Oral bioavailability may be reduced because of gut congestion. Furthermore, diuretics are 95% protein bound, hypoalbuminemia secondary to cachexia may thereby reduce the amount of the drug that reaches the kidney. Additionally, prolonged exposure to loop diuretics leads to nephron remodelling with hypertrophy of the distal tubular cells, which, in turn, may alter the diuretic response due to a compensatory increased sodium reabsorption.42
In the Spanish Heart Failure Registry (Registro de Insuficiencia Cardiaca; RICA), diuretic resistance was defined as “persistent congestion requiring hospitalisation despite adequate doses of loop diuretic (≥80 mg furosemide per day)“, and according to this definition diuretic resistance was prevalent in 21% of the admitted patients.40 The patients with diuretic resistance had lower blood pressure, more comorbidities, lower haemoglobin, lower estimated glomerular filtration rate (eGFR) and sodium, higher uric acid at admission and the patients with diuretic resistance had 37% higher risk of dying within a year.40
Worsening Renal Function
Third, a fear of worsening renal function (WRF) induced by diuretic treatment may cause clinicians to use suboptimal dosing or reduce diuretic treatment too early, before congestion relief is achieved. Traditionally, intensive diuretic treatment has been considered relatively contraindicated in HF to avoid WRF. However, mounting evidence suggests that transient WRF is not so harmful if decongestion is achieved.43,44 Importantly, renal function may improve with diuretic treatment. As part of the systemic congestion in HF with increased intraabdominal pressure and increased venous pressures, congestion also occurs in the kidneys (renal congestion).45 Elevated central venous pressure as a marker of congestion has been associated with lower eGFR; and aggressive diuretic treatment and increased urine volume in the first 24 hours of hospitalisation in HF has been associated with lower incidence of WRF.46,47
Therefore, the perceived fear of WRF may not be justified; on the contrary, it may hamper optimal congestion therapy and put patients at higher risk for adverse events.
Treatment Options
There is little guidance in choosing loop diuretics, but there is a tradition for the use of furosemide despite the fact that torsemide has increased bioavailability and a longer half-life compared with furosemide. The ongoing Torsemide comparison with Furosemide for Management of Heart Failure trial (TRANSFORM-HF trial, NCT03296813) is investigating whether torsemide is superior to furosemide in HF patients discharged from hospital. More important than diuretic agent is adequate dosing of the diuretic treatment and evaluation of the response. In the Diuretic Strategies in Patients with Acute Decompensated Heart Failure (DOSE AHF) trial comparing patients receiving IV 2.5-fold their daily oral dose with patients receiving their daily oral dose IV, the higher dose tended to be associated with favourable effects on dyspnoea relief, weight and fluid loss.31 Diuretic response is mostly assessed clinically by measuring daily change in body weight and net fluid balance. However, the correlation between weight and fluid loss is poor.37 New measures of diuretic effectiveness are called for to improve treatment strategies and tailor therapy. Measuring the concentration of sodium and chloride in urine in addition to urinary output has been suggested as a better way of evaluating congestion effect.48 The ESC recommends measuring sodium in a spot urine sample 1–2 hours following initiation of diuretics to evaluate effect on natriuresis.10
When the response to loop diuretics alone is found to be inadequate, other treatment options exist, but the level of evidence is weak. The add-on of a thiazide or thiazide-like agent, such as metolazone (sequential nephron blockade), has been associated with a higher weight and fluid loss without resulting in reduced kidney function,49 but other studies indicate increased risk of hypokalaemia and WRF.50 Mineralocorticoid receptor antagonists (MRA) may be used as diuretics and to reduce the hypokalaemic effect of loop diuretics and thiazides; however, the use in acute setting needs further investigation.51,52 In the Aldosterone Targeted Neurohormonal Combined with Natriuresis Therapy in Heart Failure trial (ATHENA), high-dose spironolactone was not associated with a reduction in natriuretic peptides or congestion relief as compared with placebo.51 However, as in many AHF trials, the control group received aggressive treatment, which may explain why intensive MRA therapy was not effective. The Acetazolamide in Decompensated Heart Failure with Volume Overload trial (ADVOR trial, NCT03505788) is currently studying whether the combination of acetazolamide and loop diuretic compared with loop diuretics alone improve diuretic response.53 A treatment option for diuretic resistance and hyponatraemia is tolvaptan, a vasopressin antagonist that enhances free water diuresis. However, convincing evidence for effectiveness is lacking.54,55 Nesiritide, a recombinant B-type natriuretic peptide, has been tested in WHF without convincing evidence.56 Serelaxin, a recombinant form of human relaxin-2 (a hormone that contributes to cardiovascular and renal adaptions during pregnancy) did not improve outcomes in acute HF.33 The new antidiabetic agent, sodium–glucose co-transporter-2 (SGLT2) inhibitor, has been shown to reduce the risk for WHF or cardiovascular death in CHF;57 future investigation will tell whether this drug has a role in the acute setting of HF.
Ultrafiltration may be used for patients who do not respond to diuretic treatment, but results regarding safety and efficacy have been unconvincing. Potential advantages of ultrafiltration were thought to be greater control over the rate and volume of fluid removal, greater net loss of sodium and less neurohormonal activation; and initial studies were promising.58 However, in the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) study, weight loss was not significantly greater with ultrafiltration compared with diuretic-based therapy, and ultrafiltration was associated with a greater increase in creatinine at 96 hours.59 Furthermore, ultrafiltration has been shown to be associated with more pronounced neurohormonal activation than diuretic treatment.60 The Aquapheresis versus Intravenous Diuretics and Hospitalization for Heart Failure (AVOID-HF) trial was stopped prematurely due to slow enrolment, but there was a non-significant trend towards longer time to first HF event in the ultrafiltration arm.61 More trials are needed to prove effectiveness for this treatment, but recently the Peripheral Ultrafiltration for the Relief from Congestion in Heart Failure (PURE-HF) trial (NCT03161158), an outcomes trial evaluating the efficacy of peripheral ultrafiltration, was also closed due to poor enrolment.
A standardised approach on diuretic dosing is difficult to propose because treatment response depends on several factors, such as body weight, kidney function, previous treatment with loop diuretics and degree of volume overload. However, in the recently published position paper on diuretic use in HF by the HFA of the ESC, a detailed algorithm for diuretic use in AHF is suggested.10 The paper emphasises the importance of early initiation of IV loop diuretics due to lowered uptake from the gut of oral medications because of gut oedema. Diuretic naive acute HF patients should receive 20–40 mg furosemide IV or an equivalent dose of other loop diuretics if kidney function is normal, otherwise a higher dose.10 Patients already on a loop diuretic admitted for AHF should receive 1–2-fold their 24-hour oral home dose intravenously. Of importance is that oral bioavailability for furosemide is highly variable (10–90%), whereas torsemide and bumetanide have a bioavailability between 80% and 100%; this needs to be taken into consideration when switching from oral to IV diuretic treatment.10,62 Additionally, evaluation of treatment effect is crucial, and diuretic doses need to be adjusted according to the response.
As outlined above, the evidence for add-on therapy when the response to loop diuretic treatment is insufficient, is weak. Detailed strategies and algorithms for treating diuretic resistance have been developed based on clinical experience and existing evidence.10,49,63 Briefly, sequential nephron blockade is mostly recommended as the second step after non-response to IV loop diuretics. Metolazone may be given in a starting dose of 2.5–5 mg once daily in combination with loop diuretics. Electrolytes should be monitored closely, and potassium substitution should be given as needed. Once a diuretic response has been generated, the frequency of metolazone treatment should be decreased or stopped completely. For patients with diuretic resistance managed in the outpatient setting, lower doses of metolazone are recommended (e.g. 2.5 mg once or twice a week). In patients with severe hyponatraemia, add-on therapy with tolvaptan may serve as an option instead of metolazone. Due to the inconclusive risk–benefit analyses, ultrafiltration should be limited to a last bail-out option if all pharmacological therapy fails.
Follow-up
As outlined above, patients discharged after an episode of AHF are at high risk for rehospitalisation. These patients should be monitored closely in the outpatient HF clinic after discharge. When a patient has reached euvolaemia, loop diuretic therapy should be reduced to the lowest dose possible to minimise WRF, neurohormonal activation and electrolyte abnormalities.10,64 However, defining the lowest effective dose is challenging. For patients already treated with diuretics before a WHF episode, a higher dose is likely needed following discharge, and accordingly, patients with no diuretic treatment before a WHF episode should be prescribed a low-dose daily loop diuretic following discharge. There is limited evidence for or against loop diuretic use, but a meta-analysis does suggest that in CHF, loop diuretics reduce the risk of death or worsening HF.65 While natriuretic peptide-guided HF therapy has not been proven effective,66 implantable haemodynamic monitoring may reduce HF hospitalisation by providing information to guide early and appropriate diuretic increases.24 At the same time, in chronic stable HF, it has been described that patients on high doses versus low doses have higher risk for mortality, sudden death and pump failure death.67 This certainly reflects confounding by severity, but loop diuretics reduce intravascular fluid and cause compensatory maladaptive neurohormonal activation and may be a risk factor, in addition to a risk marker, for worse outcomes.10 Indeed the beneficial haemodynamic effects of angiotensin receptor neprilysin inhibitors (ARNi) may reduce the need for diuretics.64 Furthermore, SGLT2 inhibitors have potent natriuretic and diuretic effects; however, the effect is dependent on volume status with less effect in euvolaemic patients, hence the treatment is associated with less adverse neurohormonal activiation.68
As hospitalised patients with WHF stabilise, evidence-based HF medication should be optimised. It has been shown that patients are more likely to be adherent to new medication initiated in hospital as compared with the outpatient clinic.69 Finally, treatment of underlying comorbidities and potential precipitation factors for the current WHF episode should be treated if possible.
Conclusion
WHF is common, underrecognised, treated too late and treated insufficiently, and associated with high risk of rehospitalisation and death. Congestion and diuretic resistance contribute to an insidious course of gradually but initially subclinical worsening HF, as well as insufficient decongestion during WHF and AHF episodes. Fear of and actual WRF limits the use of loop diuretics in WHF, but recent data suggest that residual congestion is worse than WRF, and that decongestion should be strived for, even at the risk of WRF. We believe that in contemporary clinical practice, in CHF, loop diuretics are not adjusted carefully enough, and in WHF and AHF, loop diuretic use is not aggressive enough. Improved implementation of ARNi and the introduction of SGLT2 inhibitors may alter and hopefully improve the landscape of congestion, diuretic resistance, and WHF and AHF.