










Heart failure with a preserved ejection fraction (HFpEF) comprises half of the heart failure population, and is associated with high morbidity and mortality. HFpEF is induced by multiple coexisting comorbidities, mostly of non-cardiac origin, leading to increased stiffness of the myocardial wall, diastolic dysfunction, elevated filling pressures and eventually to heart failure.1 The interplay between comorbidities greatly differs among patients; however, increasing age is a major driving force.
Many studies, researching the pathogenesis of HFpEF, focus on intracellular pathways that lead to increased cardiomyocyte stiffness.1,2 In contrast, some diseases specifically alter the extracellular matrix of the cardiac tissue, which also results in stiffening of the myocardial wall.3 Transthyretin-derived (ATTR) amyloidosis is such a disease leading to deposition of misfolded proteins in the extracellular matrix of the myocardium, and is therefore a specific extracellular cause of HFpEF.4 The aim of this review was to discuss the potential usefulness of screening for ATTR amyloidosis in the general HFpEF population, as the first treatment options for ATTR amyloidosis are now emerging.5
What is Transthyretin-derived Amyloidosis?
Two types of ATTR can be distinguished, namely the wild-type (ATTRwt) and the hereditary (ATTRv) form. The protein, transthyretin (TTR), is primarily produced in the liver, and is a transporter protein, carrying thyroxine and retinol-binding protein. A balance exists between the soluble TTR proteins in the tetrameric state and the folded monomer. In the case of the hereditary form, a mutation in the TTR gene causes the variant TTR monomers to become less stable and misfolded, leading to self-aggregation as insoluble amyloid fibrils that accumulate in the extracellular matrices of end-organs, in particular the heart, increasing myocardial stiffness and subsequently compromising normal cardiac function.6 In contrast, the pathogenesis of ATTRwt amyloid is still poorly understood; however, senescence of cardiomyocytes in combination with the cumulative exposure to oxidative stress is thought to facilitate destabilisation of the TTR tetramer, and misfolding and self-aggregation of the TTR monomer, resulting in amyloid fibrils.
The prevalence of the ATTRv amyloidosis in white people is rather low, estimated to be one per 200,000. However, ATTRv amyloidosis caused by the pathogenic V122I (TTR p.Val142Ile) variant, which almost exclusively presents with cardiac involvement, is quite common in African-Americans, with an allele frequency of 3.5%.7
The prevalence of ATTRwt amyloidosis is much higher compared with ATTRv.8 A post-mortem study demonstrated that moderate or severe ATTRwt amyloid deposition is present in 5% of HFpEF patients aged >80 years.4 In addition, González-Lopez et al. screened 120 consecutive patients with HFpEF and LV hypertrophy, and found that ATTRwt amyloidosis was present in 13% of patients with HFpEF.9 Finally, in 16% of patients with severe aorta valve stenosis, ATTRwt amyloid could be detected, which was associated with a higher mortality.10 Altogether, ATTRwt amyloidosis seems to be a common cause of HFpEF, albeit underappreciated.
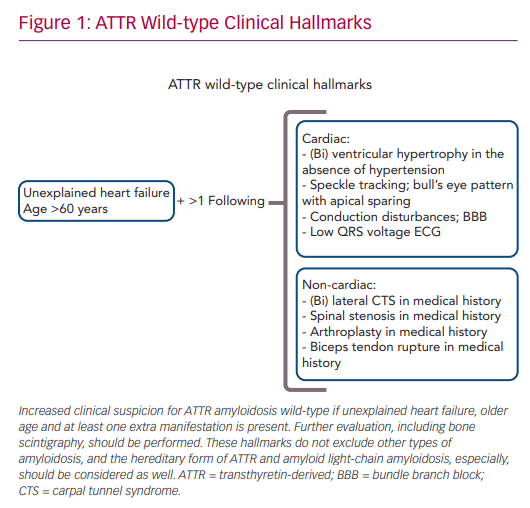
Cardiac Features of Transthyretin-derived Amyloidosis
The combination of late age of disease onset (>60 years), heart failure symptoms and left ventricular hypertrophy in the absence of hypertension increases the clinical suspicion of ATTRwt amyloidosis (Figure 1). A scintigram, as described below, is indicated to diagnose cardiac ATTR amyloidosis. Reduced exercise tolerance occurs in the early stage of the disease, whereas signs of congestive heart failure and peripheral oedema will develop later during disease progression. In approximately half of patients, AF or bundle branch blocks are present as a consequence of amyloid infiltration in atrial myocardium and the conduction system of the heart. Low QRS voltage ECGs may be present, but are less often observed.6
Non-cardiac manifestations of ATTRwt amyloidosis are frequently observed, often prior to the first cardiac manifestation.11 In half of ATTRwt patients, the presenting symptom of systemic ATTRwt amyloidosis is carpal tunnel syndrome (CTS), and it develops on average 8 years prior to the onset of the cardiomyopathy.12 Other non-cardiac manifestations include spinal canal stenosis, tendon ruptures and hip or knee arthroplasty.13,14
Therefore, (bilateral) CTS might be an early sign of ATTRwt amyloidosis, and a positive medical history for CTS should increase the clinician’s suspicion of ATTRwt amyloidosis in heart failure patients.11 Gillmore et al. proposed a staging system to further estimate the prognosis of patients with ATTR amyloid cardiomyopathy. The division is based on the plasma levels of N-terminal pro-B-type natriuretic peptide (NT-proBNP), with a cut-off value of 3,000 ng/l, and the estimated glomerular filtration rate (eGFR), with a cut-off value of 45 ml/min/1.73 m2.15 Stage I is defined by NT-proBNP levels below the cut-off value and the eGFR above the cut-off value. In stage II, either NT-proBNP is elevated or the eGFR value is decreased. Finally, stage III is defined as elevated NT-proBNP levels in combination with a decreased eGFR.15 Patients with stage I disease had a median survival of 70 months, whereas median survival in stage III was only 24 months.
How to Diagnose Transthyretin-derived Amyloidosis
Often ATTR amyloidosis is first considered in the case of the following echocardiographic findings: increased ventricular wall thickness, restrictive diastolic filling and speckle tracking that shows a bull’s eye pattern with apical sparing.6 However, as cardiac hypertrophy also often has causes other than ATTR amyloidosis, such as hypertension, aortic valve pathology and hypertrophic cardiomyopathy, the specificity of echocardiography is relatively low and thus is only useful in raising suspicion that cardiac amyloidosis is present.
An endomyocardial biopsy is the gold standard for diagnosing cardiac ATTR amyloidosis. As endomyocardial biopsy is a rather invasive procedure, an abdominal subcutaneous fat aspirate is often used instead. However, a relatively low sensitivity in detecting ATTRwt in HFpEF has been reported.16
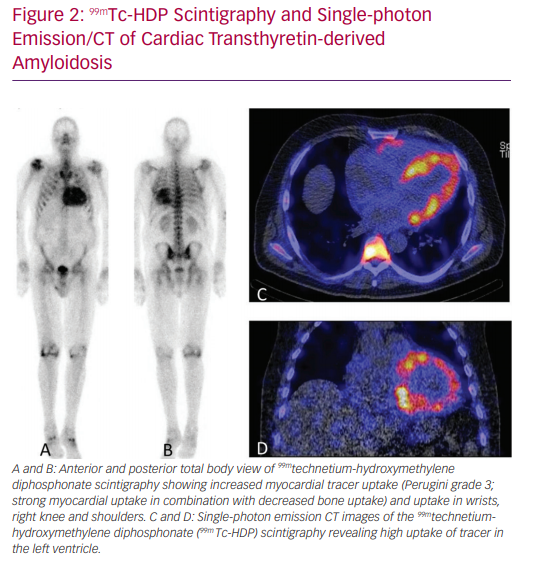
‘Bone’ scintigraphy has emerged as a very promising non-invasive alternative to diagnose ATTR amyloid cardiomyopathy (Figure 2).17 Scintigraphy detects ATTR deposition in the heart in the early stages of disease, and has a sensitivity of >99% and specificity of 86% in detecting ATTR.17 Therefore, scintigraphy is a suitable tool for the early detection of ATTRwt in the general HFpEF population.18 After a positive scintigram, genetic testing is performed to differentiate between the hereditary and wild-type form.7,17
For the hereditary form, genetic screening for the presence of a mutation in members of the family is paramount. Low- to moderate-grade tracer uptake in the heart can be observed in amyloid light-chain amyloidosis. Therefore, to rule out other forms of cardiac amyloidosis, especially with regard to in amyloid light-chain amyloidosis, further work-up is required.17
Therapeutic Implications
Until very recently, there were no proven treatment options for ATTR amyloidosis; however, several new treatment approaches have emerged that may likely change the field.5,19–22 Treatment principles are based on halting the production of the TTR protein, the stabilisation of the TTR tetramer and on the breakdown of already deposited amyloid fibrils.
Recently, the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) reported a positive effect of the TTR stabiliser, tafamidis, on the progression of ATTR amyloidosis, and consequently has become available as the first established treatment for cardiac ATTR amyloidosis.5 A reduction in all-cause mortality was observed in the group that received tafamidis over a period of 30 months. Patients with less advanced heart failure (New York Heart Association class I and II) seemed to benefit more from treatment than patients with more advanced heart failure, suggesting that stabilising TTR is useful up to a point of no return. However, one should be cautious in drawing conclusions based on subgroups of an already relatively small trial.
Another TTR tetramer stabiliser is AG10, which is capable of almost complete stabilisation of TTR.22 Although in this small study (n=49) no clinical response or effect on mortality was reported, treatment with AG10 led to normalisation of TTR plasma levels in both ATTRwt and ATTRv patients. This is promising, as decreased TTR plasma levels in untreated patients are associated with reduced survival.23 Decreased TTR plasma levels in untreated patients are deemed to reflect TTR tetramer destabilisation, leading to TTR disappearance from the circulation and ongoing accumulation of amyloid in tissue. In addition, AG10 might be more selective than tafamidis in stabilising the TTR tetramer.24
Another approach to stop amyloid accumulation is by gene silencing, which decreases the serum levels of TTR by 80–90%.25 Patisiran is a small interfering RNA drug that appeared to be safe and effective as a gene-silencing treatment option in ATTRv amyloidosis, during a treatment period of 18 months.21 Not only was disease progression of polyneuropathy halted in the treatment arm, there was also an improvement of multiple cardiac parameters, such as a reduction of the ventricular wall thickness. Collectively, this suggests that endogenous clearance of amyloid fibrils can occur to some extent if the supply of precursor protein – that is, the misfolded TTR monomer – can be significantly reduced. A similar phenomenon has been described in other systemic types of amyloidosis.26,27
Another option for gene silencing is the antisense oligonucleotide drug, inotersen. Inotersen delayed the progression of neurological disease, but as yet limited data on the effect of inotersen on cardiac parameters are available.28,29 Because of these promising results, currently both gene-silencing drugs are used in the treatment of ATTRv amyloidosis.
The last principle of treatment is the degradation of already deposited amyloid in the extracellular matrix. In one small study, 20 patients were treated with a combination of doxycycline and tauroursodeoxycholic acid for 1 year, which seemed to stabilise disease progression of primarily neurological symptoms in ATTRv patients.20 This approach is currently being investigated in patients with cardiac ATTR amyloidosis in a phase III trial that started in 2018 (NCT03481972).
Taken together, stabilisation of the TTR tetramer, to prevent dissociation into amyloidogenic monomers, is currently the leading principle of treatment in cardiac ATTR amyloidosis.14 Importantly, as described above, the treatment effect of TTR stabilisation is greatest during the early stages of disease, and is less sufficient in later disease stages. It is tempting to speculate that a combination of TTR tetramer stabilisers with small interfering RNA or future clinically applicable degraders can potentially be used to not only halt disease progression, but also lead to improvement of symptoms, particularly in patients with more advanced stages of disease.
Conclusion
In this review, we described ATTR amyloidosis to be a relatively frequent, but underdiagnosed, cause of HFpEF. The suspicion of ATTRwt amyloidosis is strong if left ventricular hypertrophy is present in the absence of hypertension and patient history reports bilateral carpal tunnel syndrome or conduction disturbances.
Currently, scintigraphy with diphosphonate or pyrophosphate tracer is a diagnostic tool that has both high specificity and sensitivity in detecting ATTR amyloid cardiomyopathy. To date, one phase III study with an ATTR stabiliser showed a beneficial effect on morbidity and mortality. Therefore, it seems that ATTR amyloidosis is becoming a treatable form of HFpEF.
In light of these results, screening for ATTR amyloidosis in the general HFpEF population with important red flags, such as left ventricular hypertrophy, conduction disturbances or a history of CTS, might be useful.